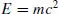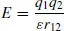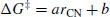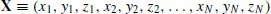Essentials of Computational Chemistry provides a balanced introduction to this dynamic subject. Suitable for both experimentalists and theorists, a wide range of samples and applications are included drawn from all key areas. The book carefully leads the reader thorough the necessary equations providing information explanations and reasoning where necessary and firmly placing each equation in context.
Preguntas frecuentes
¿Cómo cancelo mi suscripción?
Simplemente, dirígete a la sección ajustes de la cuenta y haz clic en «Cancelar suscripción». Así de sencillo. Después de cancelar tu suscripción, esta permanecerá activa el tiempo restante que hayas pagado. Obtén más información aquí.
¿Cómo descargo los libros?
Por el momento, todos nuestros libros ePub adaptables a dispositivos móviles se pueden descargar a través de la aplicación. La mayor parte de nuestros PDF también se puede descargar y ya estamos trabajando para que el resto también sea descargable. Obtén más información aquí.
¿En qué se diferencian los planes de precios?
Ambos planes te permiten acceder por completo a la biblioteca y a todas las funciones de Perlego. Las únicas diferencias son el precio y el período de suscripción: con el plan anual ahorrarás en torno a un 30 % en comparación con 12 meses de un plan mensual.
¿Qué es Perlego?
Somos un servicio de suscripción de libros de texto en línea que te permite acceder a toda una biblioteca en línea por menos de lo que cuesta un libro al mes. Con más de un millón de libros sobre más de 1000 categorías, ¡tenemos todo lo que necesitas! Obtén más información aquí.
¿Perlego ofrece la función de texto a voz?
Busca el símbolo de lectura en voz alta en tu próximo libro para ver si puedes escucharlo. La herramienta de lectura en voz alta lee el texto en voz alta por ti, resaltando el texto a medida que se lee. Puedes pausarla, acelerarla y ralentizarla. Obtén más información aquí.
¿Es Essentials of Computational Chemistry un PDF/ePUB en línea?
Sí, puedes acceder a Essentials of Computational Chemistry de Christopher J. Cramer en formato PDF o ePUB, así como a otros libros populares de Naturwissenschaften y Physikalische & theoretische Chemie. Tenemos más de un millón de libros disponibles en nuestro catálogo para que explores.
A clear definition of terms is critical to the success of all communication. Particularly in the area of computational chemistry, there is a need to be careful in the nomenclature used to describe predictive tools, since this often helps clarify what approximations have been made in the course of a modeling ‘experiment’. For the purposes of this textbook, we will adopt a specific convention for what distinguishes theory, computation, and modeling.
In general, ‘theory’ is a word with which most scientists are entirely comfortable. A theory is one or more rules that are postulated to govern the behavior of physical systems. Often, in science at least, such rules are quantitative in nature and expressed in the form of a mathematical equation. Thus, for example, one has the theory of Einstein that the energy of a particle, E, is equal to its relativistic mass, m, times the speed of light in a vacuum, c, squared,
(1.1)
The quantitative nature of scientific theories allows them to be tested by experiment. This testing is the means by which the applicable range of a theory is elucidated. Thus, for instance, many theories of classical mechanics prove applicable to macroscopic systems but break down for very small systems, where one must instead resort to quantum mechanics. The observation that a theory has limits in its applicability might, at first glance, seem a sufficient flaw to warrant discarding it. However, if a sufficiently large number of ‘interesting’ systems falls within the range of the theory, practical reasons tend to motivate its continued use. Of course, such a situation tends to inspire efforts to find a more general theory that is not subject to the limitations of the original. Thus, for example, classical mechanics can be viewed as a special case of the more general quantum mechanics in which the presence of macroscopic masses and velocities leads to a simplification of the governing equations (and concepts).
Such simplifications of general theories under special circumstances can be key to getting anything useful done! One would certainly not want to design the pendulum for a mechanical clock using the fairly complicated mathematics of quantal theories, for instance, although the process would ultimately lead to the same result as that obtained from the simpler equations of the more restricted classical theories. Furthermore, at least at the start of the twenty-first century, a generalized ‘theory of everything’ does not yet exist. For instance, efforts to link theories of quantum electromagnetics and theories of gravity continue to be pursued.
Occasionally, a theory has proven so robust over time, even if only within a limited range of applicability, that it is called a ‘law’. For instance, Coulomb’s law specifies that the energy of interaction (in arbitrary units) between two point charges is given by
(1.2)
where q is a charge, ε is the dielectric constant of a homogeneous medium (possibly vacuum) in which the charges are embedded, and r12 is the distance between them. However, the term ‘law’ is best regarded as honorific – indeed, one might regard it as hubris to imply that experimentalists can discern the laws of the universe within a finite span of time.
Theory behind us, let us now move on to ‘model’. The difference between a theory and a model tends to be rather subtle, and largely a matter of intent. Thus, the goal of a theory tends to be to achieve as great a generality as possible, irrespective of the practical consequences. Quantum theory, for instance, has breathtaking generality, but the practical consequence is that the equations that govern quantum theory are intractable for all but the most ideal of systems. A model, on the other hand, typically involves the deliberate introduction of simplifying approximations into a more general theory so as to extend its practical utility. Indeed, the approximations sometimes go to the extreme of rendering the model deliberately qualitative. Thus, one can regard the valence-shell-electron-pair repulsion (VSEPR; an acronym glossary is provided as Appendix A of this text) model familiar to most students of inorganic chemistry as a drastic simplification of quantum mechanics to permit discrete choices for preferred conformations of inorganic complexes. (While serious theoreticians may shudder at the empiricism that often governs such drastic simplifications, and mutter gloomily about lack of ‘rigor’, the value of a model is not in its intrinsic beauty, of course, but in its ability to solve practical problems; for a delightful cartoon capturing the hubris of theoretical dogmatism, see Ghosh 2003.)
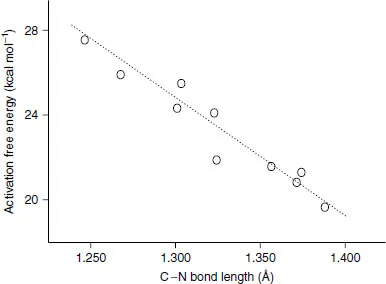
Another feature sometimes characteristic of a quantitative ‘model’ is that it incorporates certain constants that are derived wholly from experimental data, i.e., they are empirically determined. Again, the degree to which this distinguishes a model from a theory can be subtle. The speed of light and the charge of the electron are fundamental constants of the universe that appear either explicitly or implicitly in Eqs. (1.1) and (1.2), and we know these values only through experimental measurement. So, again, the issue tends to be intent. A model is often designed to apply specifically to a restricted volume of what we might call chemical space. For instance, we might imagine developing a model that would predict the free energy of activation for the hydrolysis of substituted β-lactams in water. Our motivation, obviously, would be the therapeutic utility of these species as antibiotics. Because we are limiting ourselves to consideration of only very specific kinds of bond-making and bond-breaking, we may be able to construct a model that takes advantage of a few experimentally known free energies of activation and correlates them with some other measured or predicted quantity. For example, we might find from comparison with X-ray crystallography that there is a linear correlation between the aqueous free energy of activation, ΔG‡, and the length of the lactam C–N bond in the crystal, rCN (Figure 1.1). Our ‘model’ would then be
Figure 1.1 Correlation between activation free energy for aqueous hydrolysis of β-lactams and lactam C–N bond lengths as determined from X-ray crystallography (data entirely fictitious)
(1.3)
where a would be the slope (in units of energy per length) and b the intercept (in units of energy) for the empirically determined correlation.
Equation (1.3) represents a very simple model, and that simplicity derives, presumably, from the small volume of chemical space over which it appears to hold. As it is hard to imagine deriving Eq. (1.3) from the fundamental equations of quantum mechanics, it might be more descriptive to refer to it as a ‘relationship’ rather than a ‘model’. That is, we make some attempt to distinguish between correlation and causality. For the moment, we will not parse the terms too closely.
An interesting question that arises with respect to Eq. (1.3) is whether it may be more broadly applicable. For instance, might the model be useful for predicting the free energies of activation for the hydrolysis of γ-lactams? What about amides in general? What about imides? In a statistical sense, these chemical questions are analogous to asking about the degree to which a correlation may be trusted for extrapolation vs. interpolation. One might say that we have derived a correlation involving two axes of multi-dimensional chemical space, activation free energy for β-lactam hydrolysis and β-lactam C–N bond length. Like any correlation, our model is expected to be most robust when used in an interpolative sense, i.e., when applied to newly measured β-lactam C–N bonds with lengths that fall within the range of the data used to derive the correlation. Increasingly less certain will be application of Eq. (1.3) to β-lactam bond lengths that are outside the range used to derive the correlation, or assumption that other chemical axes, albeit qualitatively similar (like γ-lactam C–N bond lengths), will be coincident with the abscissa.
Thus, a key question in one’s mind when evaluating any application of a theoretical model should be, ‘How similar is the system being studied to systems that were employed in the development of the model?’ The generality of a given model can only be established by comparison to experiment for a wider and wider variety of systems. This point will be emphasized repeatedly throughout this text.
Finally, there is the definition of ‘computation’. While theories and models like those represented by Eqs. (1.1), (1.2), and (1.3), are not particularly taxing in terms of their mathematics, many others can only be efficiently put to use with the assistance of a digital computer. Indeed, there is a certain synergy between the development of chemical theories and the development of computational hardware, software, etc. If a theory cannot be tested, say because solution of the relevant equations lies outside the scope of practical possibility, then its utility cannot be determined. Similarly, advances in computational technology can permit existing theories to be applied to increasingly complex systems to better gauge the degree to which they are robust. These points are expanded upon in Section 1.4. Here we simply close with the concise statement that ‘computation’ is the use of digital technology to solve the mathematical equations defining a particular theory or model.
With all these definitions in hand, we may return to a point raised in the preface, namely, what is the difference between ‘Theory’, ‘Molecular Modeling’, and ‘Computational Chemistry’? To the extent members of the community make distinctions, ‘theorists’ tend to have as their greatest goal the development of new theories and/or models that have improved performance or generality over existing ones. Researchers involved in ‘molecular modeling’ tend to focus on target systems having particular chemical relevance (e.g., for economic reasons) and to be willing to sacrifice a certain amount of theoretical rigor in favor of getting the right answer in an efficient manner. Finally, ‘computational chemists’ may devote themselves not to chemical aspects of the problem, per se, but to computer-related aspects, e.g., writing improved algorithms for solving particularly difficult equations, or developing new ways to encode or visualize data, either as input to or output from a model. As with any classification scheme, there are no distinct boundaries recognized either by observers or by individual researchers, and certainly a given research endeavor may involve significant efforts undertaken within all three of the areas noted above. In the spirit of inclusiveness, we will treat the terms as essentially interchangeable.
1.2 Quantum Mechanics
The postulates and theorems of quantum mechanics form the rigorous foundation for the prediction of observable chemical properties from first principles. Expressed somewhat loosely, the fundamental postulates of quantum mechanics assert that microscopic systems are described by ‘wave functions’ that completely characterize all of the physical properties of the system. In particular, there are quantum mechanical ‘operators’ corresponding to each physical observable that, when applied to the wave function, allow one to predict the probability of finding the system to exhibit a particular value or range of values (scalar, vector, etc.) for that observable. This text assumes prior exposure to quantum mechanics and some familiarity with operator and matrix formalisms and notation.
However, many successful chemical models exist that do not necessarily have obvious connections with quantum mechanics. Typically, these models were developed based on intuitive concepts, i.e., their forms were determined inductively. In principle, any successful model must ultimately find its basis in quantum mechanics, and indeed a posteriori derivations have illustrated this point in select instances, but often the form of a good model is more readily grasped when rationalized on the basis of intuitive chemical concepts rather than on the basis of quantum mechanics (the latter being desperately non-intuitive at first blush).
Thus, we shall leave quantum mechanics largely unreviewed in the next two chapters of this text, focusing instead on the intuitive basis for classical models falling under the heading of ‘molecular mechanics’. Later in the text, we shall see how some of the fundamental approximations used in molecular mechanics can be justified in terms of well-defined approximations to more complete quantum mechanical theories.
1.3 Computable Quantities
What predictions can be made by the computational chemist? In principle, if one can measure it, one can predict it. In practice, some properties are more amenable to accurate computation than others. There is thus some utility in categorizing the various properties most typically studied by computational chemists.
1.3.1 Structure
Let us begin by focusing on isolated molecules, as they are the fundamental unit from which pure substances are constructed. The minimum information required to specify a molecule is its molecular formula, i.e., the atoms of which it is composed, and the manner in which those atoms are connected. Actually, the latter point should be put more generally. What is required is simply to know the relative positions of all of the atoms in space. Connectivity, or ‘bonding’, is itself a property that is open to determination. Indeed, the determination of the ‘best’ structure from a chemically reasonable (or unreasonable) guess is a very common undertaking of computational chemistry. In this case ‘best’ is defined as having the lowest possible energy given an overall connectivity roughly dictated by the starting positions of the atoms as chosen by the theoretician (the process of structure optimization is described in more detail in subsequent chapters).
This sounds relatively simple because we are talking about the modeling of an isolated, single molecule. In the laboratory, however, we are much more typically dealing with an equilibrium mixture of a very large number of molecules at some non-zero temperature. In that case, measured properties reflect thermal averaging, possibly over multiple discrete stereoisomers, tautomers, etc., that are structurally quite different from the idealized model system, and great care must be taken in making comparisons between theory and experiment in such instances.
1.3.2 Potential Energy Surfaces
The first step to making the theory more closely mimic the experiment is to consider not just one structure for a given chemical formula, but all possible structures. That is, we fully characterize the potential energy surface (PES) for a given chemical formula (this requires invocation of the Born–Oppenheimer approximation, as discussed in more detail in Chapters 4 and 15). The PES is a hypersurface defined by the potential energy of a collection of atoms over all possible atomic arrangements; the PES has 3N − 6 coordinate dimensions, where N is the number of atoms ≥3. This dimensionality derives from the three-dimensional nature of Cartesian space. Thus each structure, which is a point on the PES, can be defined by a vector X where
(1.4)
and xi, yi, and zi are the Cartesian coordinates of atom i. However, this expression of X does not uniquely define the structure because it involves an arbitrary origin. We can reduce the dimensionality without affecting the structure by removing the three dimensions associated with translation of the structure in the x, y, and z directions (e.g., by insisting that the molecular center of mass be at the origin) and removing the three dimensions associated with rotation about the x, y, and z axes (e.g., by requiring that the principal moments of inertia align along those axes in increasing order).
A different way to appreciate this reduced dimensionality is to imagine constructing a structure vector atom by atom (Figure 1.2), in which case it is most convenient to imagine the dimensions of the PES being internal coordinates (i.e., bond lengths, valence angles, etc.). Thus, choice of the first atom involves no degrees of geometric freedom – the atom defines the origin. The position of the second atom is specified by its distance from the first. So, a two-atom system has a single degree of freedom, the b...