
eBook - ePub
Intracranial Gliomas Part II - Adjuvant Therapy
- 252 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Intracranial Gliomas Part II - Adjuvant Therapy
About this book
Treatment of patients with intracranial gliomas, especially high-grade neoplasms, usually requires postoperative adjuvant therapy. Significant progress in the understanding of tumor biology, technological advances in irradiation delivery, and development of novel antitumor drugs have led to an expansion of the therapeutic arsenal in neuro-oncology. This publication provides a unique review of the various options for adjuvant therapy. Special emphasis is on current evidence-based treatment standards and guidelines, and on perspectives of further improvement in long-term outcomes. Chapters review the histopathological and molecular features of gliomas and describe basic principles and clinical results of fractionated radiotherapy, stereotactic radiosurgery, brachytherapy, use of radiosensitizers, systemic chemotherapy and antiangiogenic therapy. Particular attention is paid to treatment of pediatric patients and to physical and psychological rehabilitation and supportive care at the end of life. This book and its accompanying volumes are mainly directed at neuro-oncologists, radiation oncologists, and other clinicians treating patients with brain tumors.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Chernov MF, Muragaki Y, Kesari S, McCutcheon IE (eds): Intracranial Gliomas. Part II – Adjuvant Therapy. Prog Neurol Surg. Basel, Karger, 2018, vol 31, pp 1–37 (DOI: 10.1159/000466835)
______________________
Pathology and Genetics of Gliomas
Takashi Komoria · Yoshihiro Muragakib, c · Mikhail F. Chernovb, c
aDepartment of Laboratory Medicine and Pathology (Neuropathology), Tokyo Metropolitan Neurological Hospital, and bFaculty of Advanced Techno-Surgery and cDepartment of Neurosurgery, Tokyo Women’s Medical University, Tokyo, Japan
______________________
Abstract
Current World Health Organization (WHO) classification of the neuroepithelial tumors is cell lineageoriented and based on a presumed developmental tree of the central nervous system (CNS). It defines three main groups of gliomas, namely astrocytomas, oligodendrogliomas, and ependymomas, and additionally presumes their 4-tiered histopathological grading (WHO grades I to IV). Nevertheless, the impact of tumor pathology on clinically related parameters may be frequently much better predicted by genetics, than by histological appearance of the lesion. Recent studies have revealed several major molecular alterations typical for different types of neoplasms, such as IDH1/IDH2 mutations in diffusely infiltrating gliomas, mutations of TP53 and ATRXin astrocytomas, 1p/19q co-deletion in oligodendrogliomas, mutations of TERT promoter in oligodendrogliomas and IDH wild-type glioblastomas, and mutations or fusions of BRAF in circumscribed astrocytomas, particularly in children. Identification of those and several other genetic abnormalities in the tumor is clinically important and may help clinicians to determine proper treatment strategy and to predict prognosis. Therefore, the updated WHO classification of CNS tumors (2016) considers not only phenotype, but also some genetic characteristics of gliomas.
© 2018 S. Karger AG, Basel
Introduction
In 1926 Percival Bailey and Harvey Cushing presented the first classification of gliomas based on the presumed developmental tree of the central nervous system (CNS) [1]. This cell lineage-oriented concept has long been a central basis of the World Health Organization (WHO) classification of CNS tumors. Despite regular updates according to new information on histology, immunohistochemistry (IHC), and ultrastructure, even in its 4th edition published in 2007 [2, 3] this classification outlined only the histogenetic profile of each neoplasm.
Nonetheless, it is now clear that neuroepithelial tumors have the potential to differentiate beyond the presumed developmental tree of the CNS and that various lineages of differentiation do not necessarily correlate with the biological behavior of the mass lesion [4]. Moreover, characterization of gliomas based on descriptive histological criteria has been always accompanied by more or less considerable interobserver variability, especially in cases of mixed and heterogeneous neoplasms, caused by subjective interpretation of the microscopic tumor appearance and/or small volume of the biopsy material [5, 6]. Finally, the impact of tumor pathology on clinically related parameters (e.g., response to therapy or survival) may be frequently much better predicted by genetics, than by histological characteristics [5, 7–9]. Therefore, solely cell lineage-oriented classification of gliomas appears to no longer be rational and an alternative approach for typing and grading of brain tumors based on molecular information has thus been sought [4].
To discuss the incorporation of genetic data into the next edition of the WHO classification, a consensus meeting of neuropathologists with an expertise in molecular diagnosis was held in May 2014 in Haarlem, the Netherlands, under the sponsorship of the International Society of Neuropathology (ISN) [10]. Established “ISN-Haarlem consensus guidelines” were reflected in part in the updated WHO classification of CNS tumors (2016) [11], which considers not only phenotype, but also some genetic fingerprints of the neoplasms. This chapter presents contemporary concepts of the histopathological classification of gliomas based on the current WHO criteria and their possible future modification reflecting diagnostic, prognostic, and predictive values of major molecular alterations in tumors.
Contemporary Histopathological Classification of Gliomas
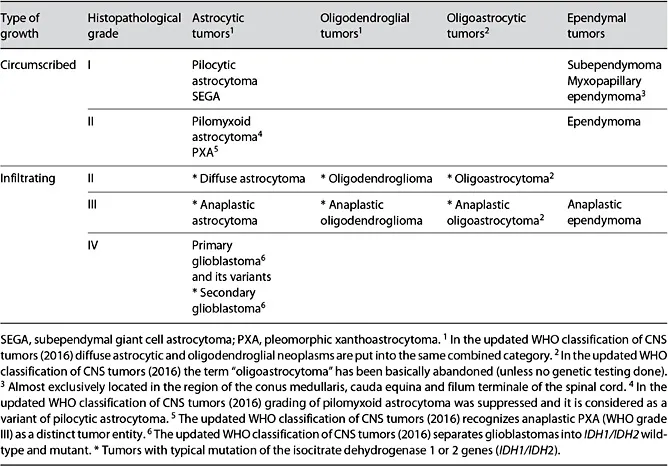
According to the 4th edition of the WHO classification of CNS tumors (2007) [2, 3] the vast majority of gliomas comprises four histological groups, namely astrocytomas, oligodendrogliomas (OD), mixed oligoastrocytomas, and ependymomas, according to their microscopic morphological similarities with the normal cellular counterparts (Table 1). Additionally, this classification presumed histopathological grading of the tumor (WHO grades I–IV). In general, typing of the neoplasm is directed at the recognition of its biological origin, while grading determines a stage in the malignant progression [12].
Nevertheless, the updated WHO classification of CNS tumors (2016) [11] for the first time considers the presence of some genetic alterations in diffuse gliomas, mainly mutations of the isocitrate dehydrogenase 1 and 2 genes (IDH1/IDH2) and combined complete loss of the chromosomal arms 1p and 19q (1p/19q co-deletion), which are incorporated into the lesion name (Fig. 1). Thus for pathological characterization of the neoplasm molecular testing is considered mandatory. If it is not available or cannot be fully performed, an NOS (not otherwise specified) definition is applied. Notably, during establishment of diagnosis for diffuse astrocytic and oligodendroglial tumors the genotype trumps the histological phenotype. Additionally, the updated WHO classification of CNS tumors (2016) [11] has made several changes in designated tumor entities, variants, and patterns.
Table 1 Framework for pathological classification of gliomas according to the 4th edition (2007) of the WHO classification of CNS tumors (modified from Komori [4])
Astrocytomas (WHO Grades I–IV)
Astrocytes are multipolar, star-like cells of the CNS with an eosinophilic cytoplasm and cytoplasmic processes. The term “astrocytoma” widely applies to tumors that exhibit astrocytic differentiation. Microscopically these lesions appear as hypercellular area of neoplastic cells with irregular, elongated hyperchromatic nuclei, a high degree of fibrillarity, intermixture with normal brain elements, and the frequent formation of secondary structures around neurons, blood vessels and beneath the pia mater [13, 14]. Nuclear hyperchromasia and enlargement as well as cellular crowding and clustering may distinguish neoplastic and reactive astrocytes [14]. On IHC, glial fibrillary acidic protein (GFAP) is a hallmark of astrocytic differentiation; however, it is obviously not neoplasm-specific and is less expressed in undifferentiated examples.
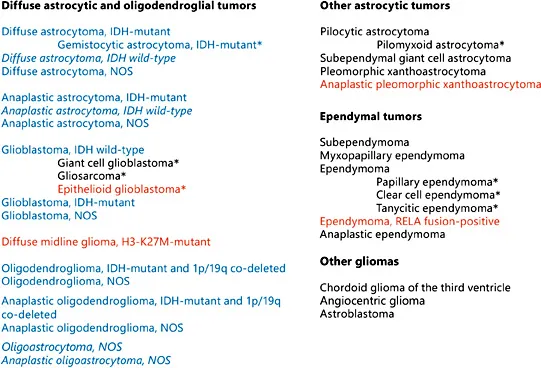
Fig. 1. Nomenclature for gliomas according to the updated WHO classification of CNS tumors (2016) [11]. NOS, not otherwise specified (no genetic testing done). Italic, provisional entities; blue, new genetic-based nomenclatures; red, new entities or variants. * A variant.
The group of astrocytomas includes both circumscribed and infiltrating lowgrade (LGG) and high-grade (HGG) gliomas. Circumscribed lesions correspond to unique tumor types, such as pilocytic astrocytoma (PA; WHO grade I), subependymal giant cell astrocytoma (SEGA; WHO grade I), and pleomorphic xanthoastrocytoma (PXA; WHO grade II), which mainly occur in children and young adults and are generally associated with a more or less indolent clinical course. Diffusely infiltrating astrocytomas are divided into 3 main types, namely diffuse astrocytoma (DA; WHO grade II), anaplastic astrocytoma (AA; WHO grade III) and glioblastoma multiforme (GBM; WHO grade IV). Although the majority of infiltrating astrocytomas show fibrillary structure, there is a large morphological heterogeneity, including gemistocytic, small cell, granular cell, giant cell and epithelioid subtypes. With few exceptions these variants do not pose a u...
Table of contents
- Cover Page
- Front Matter
- Pathology and Genetics of Gliomas
- Fractionated Radiotherapy of Intracranial Gliomas
- Stereotactic Radiosurgery in the Multimodality Management of Residual or Recurrent Glioblastoma Multiforme
- Stereotactic Radiosurgery of Intracranial Low-Grade Gliomas
- Brachytherapy of Intracranial Gliomas
- Irradiation of Intracranial Gliomas in Children
- Role of Radiosensitizers in Radiation Treatment of Gliomas
- Chemotherapy of High-Grade Astrocytomas in Adults
- Chemotherapy of Diffuse Astrocytoma (WHO grade II) in Adults
- Chemotherapy of Oligodendrogliomas
- Chemotherapy of Intracranial Gliomas in Children
- Perspectives of Personalized Chemotherapy of Gliomas Based on Molecular Tumor Profiling
- Antiangiogenic Therapy of High-Grade Gliomas
- Search for More Effective Chemotherapeutic Regimens for Gliomas: Challenges and Hopes
- Physical and Psychological Rehabilitation of Patients with Intracranial Glioma
- Palliative and Supportive Care of Patients with Intracranial Glioma
- Author Index
- Subject Index
- Back Cover Page
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Intracranial Gliomas Part II - Adjuvant Therapy by M. F. Chernov,Y. Muragaki,S. Kesari,I. E. McCutcheon,M.F., Chernov,Y., Muragaki,S., Kesari,I.E., McCutcheon, Konstantin V. Slavin,Konstantin V., Slavin in PDF and/or ePUB format, as well as other popular books in Medicine & Neurology. We have over 1.5 million books available in our catalogue for you to explore.