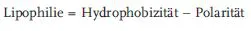Durch die jüngste Änderung der Approbationsordnung für Apotheker hat die Bedeutung der Biopharmazie in Forschung und Lehre weiter zugenommen. In diesem vollständig neu konzipierten Lehr- und Handbuch behandeln renommierte Autoren sämtliche Themen der Biopharmazie entsprechend den neuen Anforderungen. Aktuell und übersichtlich, richtet sich das Grundlagenwerk an Pharmazeuten in Wissenschaft und Industrie, aber auch an Studenten, die besonders von den integrierten Übungsteilen profitieren.
Die Hauptkapitel zu den Grundlagen der Physiologie und Pharmakokinetik werden ergänzt durch Abschnitte zu Anwendungen der Biopharmazie in der Arzneimittelentwicklung und in der Klinik. Zu topaktuellen Themen wie Prodrugs und Drug Targeting referiert der Band den Stand der Forschung. Für Praktiker hält er außerdem ein Kapitel zu Computerprogrammen in der Biopharmazie bereit. Auch Studenten hat das Buch eine Menge zu bieten: Zahlreiche Übungsaufgaben sowie Verständnisfragen mit den dazugehörigen Antworten erlauben eine effektive Lernkontrolle und damit eine optimale Prüfungsvorbereitung. Der Band folgt der Terminologie der Europäischen Pharmakopöe 2001. Das ausführliche Glossar enthält mehr als 100 Begriffe. Außerdem werden über 130 pharmakokinetische Abkürzungen und Symbole erklärt, so dass das Buch auch als Nachschlagewerk genutzt werden kann. Das Autorenteam hat sich einiges vorgenommen: Ihr Buch soll das Referenzwerk der Biopharmazie werden, für Studenten und Dozenten der Pharmazie, Pharmazeuten und Pharmakologen ebenso wie für Praktiker in der Pharmaindustrie.
Angewandte Biopharmazie und Pharmakokinetik in der Arzneimittelentwicklung
5.1 Physikalisch-chemische Grundlagen der Absorption: Biopharmazeutisch relevante Wirkstoff-Eigenschaften
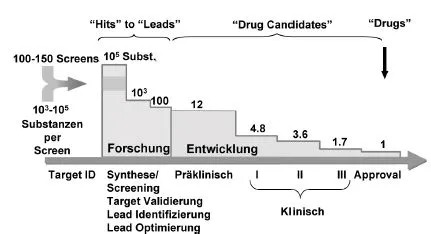
Für die Entwicklung neuer Arzneistoffe spielt die Optimierung ihrer pharmakokinetischen Eigenschaften neben ihrem pharmakodynamischen Profil (s. Kap. 6.1) eine immer wichtigere Rolle. Unter Verwendung von kombinatorischer Chemie und in-vitro Testsystemen an isolierten, teilweise klonierten pharmakologischen Rezeptoren kann heute unter Anwendung von „High-Throughput Screening-Methoden“ (HTS-Methoden) eine große Anzahl von potenziellen Wirkstoffen synthetisiert und auf in-vitro Rezeptoraffinität getestet werden. Trotz dieser Anstrengungen ist jedoch die Anzahl der entwickelbaren Substanzen und derjenigen, aus denen schließlich ein Arzneimittel wird, gemessen an der Zahl der zu testenden Substanzen recht bescheiden (Abb. 5.1).
Die Entwicklung in der Laborautomation wurde dabei insbesondere durch Fortschritte auf den Gebieten der Laborrobotersysteme (z. B. Sagian®, Staccato® und Tecan®) beschleunigt. Solche Systeme übernehmen z. B. die Handhabung der in-vitro Inkubationsprozesse in Plattensystemen, in denen von 96 über 384 und 864 bis zu 1536 isolierte Reaktionskammern (so genannte Wells) untergebracht sind. Weiterhin werden die Roboter zur Organisation und Durchführung von Inkubationen, Plattenauswertungen (Readers), Filtrationen und Flüssigkeits-Handlingprozessen, z. B. der Herstellung von Verdünnungsreihen (Burnbaum, 1997; Rogers, 1997) eingesetzt. Dazu gesellt sich jeweils eine an die Aufgabenstellung angepasste Software, welche die Datenauswertung, Archivierung und z. T. Modellierung übernimmt (Bioinformatik). In den Anfängen wurden HTS-Methoden vor allem zur pharmakologischen Identifizierung und Selektion von Substanzen gegenüber bestimmten pharmakologischen Rezeptoren eingesetzt. Dabei wurden diejenigen Wirkstoffe als besonders viel versprechend identifiziert, die in besonders geringer Konzentration und mit hoher Affinitat an eine bestimmte Rezeptorpräparation binden. Häufig handelt es sich dabei um Moleküle, die ein hohes Molekulargewicht (zahlreiche funktionelle Gruppen = hohe Wahrscheinlichkeit der Interaktion mit den funktionellen Gruppen am Rezeptor) und eine große Lipophilie (hydrophobe Wechselwirkung mit dem Rezeptorprotein verstärkt die Bindungsaffinität) aufweisen. In Tab. 5.1 sind charakteristische Veränderungen in den Molekulargewichten und in der Lipophilie von neu synthetisierten Verbindungen in der prä- und post-HTS-Ära aufgelistet.
Abb. 5.1 Traditionelle pharmazeutische Forschung und Entwicklung unterliegt hohem Risiko. Es wird der Selektionsprozess über den Zeitraum von der Substanzfindung bis zur Zulassung eines neuen Arzneimittels (Approval) deutlich. Unter dem waagerechten Pfeil, beginnend mit der Identifizierung des Zielrezeptors und einer damit beeinflussbaren Krankheit, sind die verschiedenen Phasen der präklinischen und klinischen Forschung und Entwicklung und die Anzahl der Wirksubstanzen, die in die jeweils nächste Stufe gelangen, angegeben.
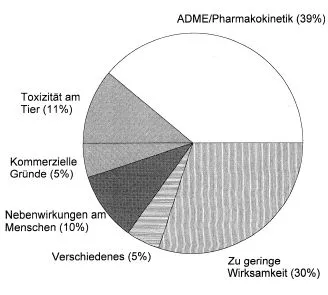
Diese Entwicklung und Selektion geeigneter Wirkstoffkandidaten in-vitro und auf der alleinigen Basis der pharmakologischen Potenz der Verbindung war jedoch mit dem Nachteil behaftet, dass biopharmazeutische Substanzeigenschaften häufig nicht mitbedacht und -optimiert wurden, da diese überwiegend in-vivo, d.h. am lebenden Organismus erhoben werden mussten. In-vivo Untersuchungen sind nach wie vor relativ arbeitsaufwendig, teils in der Gesellschaft umstritten (Tierversuche) und an aufwendige ethische Genehmigungsverfahren geknüpft und damit auch nur mit relativ begrenztem Substanzdurchsatz durchzuführen. In Abb. 5.2 sind die unterschiedlichen Ursachen dargelegt, aufgrund derer eine Gruppe von 198 untersuchten Wirkstoffen in unterschiedlichen Phasen der Forschung und Entwicklung vor der Zulassung zurückgezogen werden mussten.
Es zeigt sich, dass die Limitationen in pharmakokinetischen Teilprozessen wie Absorption, Distribution, Metabolismus und Exkretion (ADME) als wesentliche Elemente des LADME-Modells (L = Liberation) in 39% der Fälle als Ursachen für das Abbrechen einer Substanzentwicklung identifiziert wurden. Typische Erkennungsmerkmale dieser pharmakokinetisch problematischen Substanzen sind in Tab. 5.2aufgezählt.
Tab. 5.1: Änderung der physikochemischen Eigenschaften synthetisierter, nicht registrierter Verbindungen in der prä- (1986) und post-HTS Ära (1994). 90-tes Perzentil heißt dass unterhalb des Wertes 90% aller Einzelwerte liegen, analog sind die Angaben für 75tes und 50-tes Perzentil zu interpretieren (nach Lipinski et al., 1997). M logP ist der nach Moriguchi berechnete logP-Wert (Moriguchi et al., 1992)
Abb. 5.2 Risiken bei der Arzneimittelwirkung. Die ADME-Problematik trägt zu 39% zum Abbruch der Entwicklung einer neuen Wirksubstanz (NCE; New Chemical Entity) bei. Untersucht wurden 198 Fälle. Werden die Wirkstoffe aus der Gruppe der Antiinfektiva aus dem Datensatz entfernt, so sinkt der Anteil der pharmakokinetischen Ursachen für den Abbruch einer Entwicklung auf 7%. (nach Kennedy, 1997).
Tab. 5.2: Mögliche Probleme der Pharmakokinetik (ADME) bei neu synthetisierten Verbindungen
Häufig werden derart fehlerbehaftete Entwicklungsprojekte erst (zu) spät entdeckt, d.h. zu einem Zeitpunkt, bei dem die klinische Entwicklung einer Substanz bereits fortgeschritten ist und viel Geld in die Neuentwicklung investiert wurde. Daher ist es seit einigen Jahren erklärtes Ziel, Problemsubstanzen bereits in einer frühen Phase des Entdeckungs- und Entwicklungsprozesses zu identifizieren und auszusortieren bzw. zu optimieren („Early-ADM“). Hierbei werden, analog den pharmakologischen High-throughput Verfahren, Methoden entwickelt, die das pharmazeutische, pharmakokinetische und biopharmazeutische Verhalten einer Substanz im Organismus möglichst präzise nachahmen oder vorhersagen können. Falls eine derartige Vorhersage nicht möglich sein sollte, so wird häufig auch eine Rangordnung (Ranking) von Substanzkandidaten aus einer Gruppe akzeptiert, um nur die Substanzen mit den besten in-vitro Eigenschaften weiter zu verfolgen. Dabei sind als Voraussetzungen für die Eignung einer Methode (1) ein geringer Substanzbedarf, (2) die rasche und möglichst automatisierbare Bestimmung bestimmter Substanzparameter und (3) der Nachweis, dass jene charakteristischen Substanzeigenschaften innerhalb bestimmter Grenzwerte für pharmakokinetische Teilprozesse von Bedeutung sind. Für physikochemische Substanzparameter wie z. B. pKa-Wert, log P-Wert (log D-Wert), Löslichkeit aber auch für pharmakokinetische Teilprozesse wie z. B. Absorption, metabolische Stabilität, Toxizität sind derartige Screeningsysteme etabliert. Ein weiteres interessantes Verfahren zur Erhöhung des Substanzdurchsatzes ist das Cassetten-Dosierverfahren. Damit wird in einem in-vitro Test, z. B. einer Permeabilitätsuntersuchung oder einer in-vivo Pharmakokinetik-Studie anstelle von jeweils nur einer Substanz pro Versuch ein Gemisch aus 5 bis 10 Substanzen gleichzeitig untersucht. Voraussetzung für die Anwendung dieser Methode ist das Vorhandensein einer geeigneten Analytik, mit der jede der Einzelsubstanzen aus der Mischung quantifiziert werden kann. Üblicherweise werden heute dazu selektive Bestimmungsmethoden wie z. B. massenspektrometrische Verfahren (z. B. LC-MS/MS) eingesetzt. Eine weitere Voraussetzung für die Äquivalenz von Ergebnissen aus Cassetten-Dosierverfahren im Vergleich zu herkömmlichen Bestimmungsmethoden ist die Abwesenheit von Interaktionen. Diesem Punkt versucht man dadurch zu begegnen, dass die gewählten Dosen bzw. Substanzkonzentrationen möglichst gering gewählt werden, andernfalls besteht die Gefahr einer Sättigung aktiver Prozesse. Ferner sollten Standardsubstanzen als Qualitätskontrollproben dem Gemisch beigefügt werden, von denen man die biologische Antwort bereits im Vorfeld kennt. Sowohl im Fall von in-vitro Permeationsuntersuchungen (Tannergren et al., 2001) als auch in-vivo Studien (Gaviraghi et al., 2001) haben sich Cassetten-Dosierverfahren als sinnvoll erwiesen.
Die Frage, wie rasch und in welchem Umfang ein extrasystemisch verabreichter Wirkstoff absorbiert und im Blutkreislauf zugänglich wird und wie er sich dann weiter verhält, ist von einer Vielzahl von Faktoren abhängig, die in physikalisch-chemische und biochemische Faktoren unterteilt werden können. In Abb. 5.3 sind einige dieser Faktoren, bezogen auf die Absorption am Beispiel der peroralen Applikation zusammengefasst.
Abb. 5.3 Physiologisch-biochemische und physikochemisch/formulierungstechnische Faktoren, die die Absorption von Arzneistoffen aus dem Gastrointestinaltrakt beeinflussen.
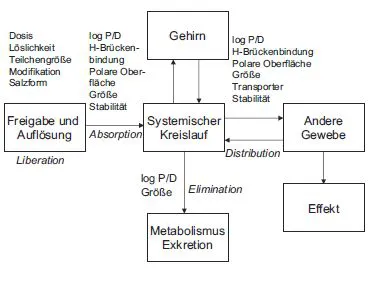
Die hauptsächlichen physikalisch-chemischen Eigenschaften, die die pharmakokinetischen Eigenschaften von Wirkstoffen beeinflussen, sind ihre Lipophilie und das Ausmaß ihrer Ionisation in der physiologischen Umgebung, ihre Löslichkeit in wässrigem Milieu, die Tendenz, mit Wassermolekülen Bindungen einzugehen (z. B. Wasserstoff-Brückenbindungen), die Stabilität in biologischen Medien und ihre Diffusionsfähigkeit aufgrund der Molekülgröße. Für alle diese Eigenschaften gibt es fließende Grenzen, nach deren Überschreiten der ungestörte Ablauf bestimmter pharmakokinetischer Teilprozesse, z. B. der Absorption, eher unwahrscheinlich wird. Dabei sollte aber keiner dieser biopharmazeutischen Wirkstoffparameter isoliert betrachtet werden, da die Optimierung des einen Teilprozesses eine Verschlechterung eines zweiten Prozesses mit sich bringen kann. Der Zusammenhang zwischen physikochemischen Substanzparametern und den Vorgängen der LADME-Prozesse ist in Abb. 5.4 gezeigt.
5.1.1 Lipophilie, pKa-Wert und Ionisation
Die Lipophilie ist ein Maß für die Neigung eines Moleküls, sich in einer lipophilen Phase („Fettphas“) bevorzugt anzureichern. Sie ist aus zwei Komponenten zusammengesetzt (Testa et al., 1996):
Als Hydrophobizität bezeichnet man dabei die Tendenz unpolarer Moleküle oder Molekülgruppen, sich in wässrigen Lösungen zusammenzulagern. Dies ist darauf zurückzuführen, dass die ausgeprägten intermolekularen Bindungen (Wasserstoffbrücken) zwischen den Wassermolekülen, welche zu der bekannten Cluster-Struktur des Wassers führen, die Tendenz haben, unpolare Moleküle auszuschließen. Polarität hingegen ist ein Maß für die Anzahl und Stärke der Wasserstoffbrückenbindungen, die ein Molekül mit den umgebenden Wassermolekülen eingeht. Dafür ist in erster Linie die Anzahl der polaren funktionellen Molekülgruppen verantwortlich, wie z. B. Amine, Amide, Ether, Ketone, Nitrile, Carboxylate, Sulfone, Sulfoxide.
Abb. 5.4 Das LADME-Modell und biopharmazeutisch relevante Substanzparameter, die einen Einfluss auf die einzelnen Prozesse haben können.
Für die Bestimmung der Lipophilie gibt es unterschiedliche Verfahren. Besonders gebräuchlich ist die Messung der Verteilung zwischen einer lipophilen und einer wässrigen Phase. Das Prinzip der Methode ist in Kapitel 4.5.1.1 dargestellt. Der dekadische Logarithmus des Quotienten der Gleichgewichtskonzentration einer Substanz zwischen lipophiler Phase und Puffer wird auch als log D-Wert bezeichnet. Als Faustregel gilt, dass Substanzen mit einem log DpH 7,4 < 0 nur schwer in der Lage sind, Lipidmembranen zu überwinden. n-Octanol ist die am häufigsten verwendete lipophile Phase, obwohl dieses System sich nicht ideal bezüglich der der Verteilung (Nernst-Bedingungen) verhält. So kann n-Octanol z. B. durch die alkoholische funktionelle Gruppe Wasserstoffbrückenbindungen mit gelösten Substanzen eingehen. Darüber hinaus sind im Gleichgewicht mit Wasser vier Volumenprozente Wasser im n-Octanol enthalten. Damit bleibt eine der Voraussetzungen der Methode, die prinzipielle Nichtmischbarkeit der Phasen, teilweise unerfüllt. Neben n...
Inhaltsverzeichnis
Cover
Title Page
Copyright
Vorwort
Geleitwort
I: Grundlagen der Biopharmazie
II: Anwendungen der Biopharmazie
Anhang I – Symbole Pharmakokinetik und Proteinbindung
Anhang II – Lösungen zu den Fragen
Register
Häufig gestellte Fragen
Ja, du kannst dein Abo jederzeit über den Tab Abo in deinen Kontoeinstellungen auf der Perlego-Website kündigen. Dein Abo bleibt bis zum Ende deines aktuellen Abrechnungszeitraums aktiv. Erfahre, wie du dein Abo kündigen kannst
Nein, Bücher können nicht als externe Dateien, z. B. PDFs, zur Verwendung außerhalb von Perlego heruntergeladen werden. Du kannst jedoch Bücher in der Perlego-App herunterladen, um sie offline auf deinem Smartphone oder Tablet zu lesen. Erfahre, wie du Bücher herunterladen kannst, um sie offline zu lesen
Perlego bietet zwei Abopläne an: Elementar und Erweitert
Elementar ist ideal für Lernende und Profis, die sich mit einer Vielzahl von Themen beschäftigen möchten. Erhalte Zugang zur Basic-Bibliothek mit über 800.000 vertrauenswürdigen Titeln und Bestsellern in den Bereichen Wirtschaft, persönliche Weiterentwicklung und Geisteswissenschaften. Enthält unbegrenzte Lesezeit und die Standardstimme für die Funktion „Vorlesen“.
Pro: Perfekt für fortgeschrittene Lernende und Forscher, die einen vollständigen, uneingeschränkten Zugang benötigen. Schalte über 1,4 Millionen Bücher zu Hunderten von Themen frei, darunter akademische und hochspezialisierte Titel. Das Pro-Abo umfasst auch erweiterte Funktionen wie Premium-Vorlesen und den Recherche-Assistenten.
Beide Abopläne sind mit monatlichen, halbjährlichen oder jährlichen Abrechnungszyklen verfügbar.
Wir sind ein Online-Lehrbuch-Abo, bei dem du für weniger als den Preis eines einzelnen Buches pro Monat Zugang zu einer ganzen Online-Bibliothek erhältst. Mit über 1 Million Büchern zu über 990 verschiedenen Themen haben wir bestimmt alles, was du brauchst! Erfahre mehr über unsere Mission
Achte auf das Symbol zum Vorlesen bei deinem nächsten Buch, um zu sehen, ob du es dir auch anhören kannst. Bei diesem Tool wird dir Text laut vorgelesen, wobei der Text beim Vorlesen auch grafisch hervorgehoben wird. Du kannst das Vorlesen jederzeit anhalten, beschleunigen und verlangsamen. Erfahre mehr über die Funktion „Vorlesen“
Ja! Du kannst die Perlego-App sowohl auf iOS- als auch auf Android-Geräten nutzen, damit du jederzeit und überall lesen kannst – sogar offline. Perfekt für den Weg zur Arbeit oder wenn du unterwegs bist. Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Ja, du hast Zugang zu Biopharmazie von Gert Fricker,Peter Langguth,Heidi Wunderli-Allenspach im PDF- und/oder ePub-Format sowie zu anderen beliebten Büchern aus Naturwissenschaften & Industrielle & technische Chemie. Aus unserem Katalog stehen dir über 1 Million Bücher zur Verfügung.