
eBook - ePub
GMP-Qualifizierung und Validierung von Wirkstoffanlagen
Ein Leitfaden für die Praxis
- German
- ePUB (handyfreundlich)
- Über iOS und Android verfügbar
eBook - ePub
Über dieses Buch
Unter Validierung bzw. Qualifizierung versteht man die Beweisf hrung, dass Verfahren, Prozesse, Ausr stungsgegenst nde, Materialien, Arbeitsg nge oder Systeme tats chlich zu den erwarteten Ergebnissen f hren. Betroffen sind alle Unternehmen, die Rohstoffe, Halbfertig- oder Fertigprodukte f r medizinische Ger te, Pharmazeutika, Diagnostika, Lebensmittel herstellen. Ebenso sind Labore betroffen, die Dienstleistungen anbieten, deren Ergebnisse direkt in den Herstellungsprozess einflie en. Dieses Buch liefert "harte Fakten" hinsichtlich der Durchf hrung (How to do) von praxiserprobten Qualifizierungs- und Validierungsma nahmen - ein "Must have" f r Wirkstoff- und Arzneimittelhersteller sowie deren Zulieferer. Der deutsche Titel zur Validierung und Qualifizierung
375,005 Studierende vertrauen auf uns
Zugang zu über 1 Million Titeln zu einem fairen monatlichen Preis.
Mit unseren Lerntools kannst du noch effizienter lernen.
Information
1
Einführung
Arzneimittel sind aus unserem täglichen Leben nicht mehr wegzudenken. Keine Errungenschaft hat die Gesellschaft je so umfassend geprägt und so wesentlich verändert, wie die in vielfältigsten Formen für unterschiedlichste Zielrichtungen und Anwendungsfälle eingesetzten pharmazeutischen Produkte. Ob zum Zwecke der Heilung oder um Schmerzen zu lindern, ob zur Vorbeugung und Kräftigung oder nur, um der Schönheit zu dienen. Jene kleinen Pillen, die schnell und einfach geschluckt dem Magendruck entgegenwirken, die Spritze beim Arzt, die vorbeugend jenen notwendigen Impfschutz verleiht, der beruhigt der Schnupfenzeit entgegensehen lässt. Die Wunddesinfektion, die verhindert, dass die kleinen Mikroben ihr Unwesen treiben – wer wollte, wer könnte auf all diese Errungenschaften verzichten. Die Entwicklungen haben Jahre gedauert, aber auch Jahre gebracht – Lebensjahre in einer immer älter werdenden Gesellschaft. Ohne Medizin und Arzneimittel nicht denkbar.
Arzneimittel sind nicht neu. Von alters her beschäftigen sich die Menschen sehr intensiv mit den heilenden, den stärkenden und lebensverlängernden Wirkungen von Extrakten und Tinkturen, ursprünglich ausschließlich aus dem Reich der Natur gewonnen. Studiert werden die Auswirkungen auf Seele und Geist, auf das psychische und physische Wohlbefinden des Menschen mit dem Ziel, diesem das Erdendasein so lang und so angenehm als möglich zu gestalten. Mit dem Studium der Substanzen, dem Eindringen in die Welt der Moleküle und dem gesamten sich offenbarenden Reich der Chemie schienen den Möglichkeiten auch kaum noch Grenzen gesetzt zu sein. Nur die Errungenschaften der modernen Biotechnologie konnten diesem Thema nochmals einen gewaltigen Schub verleihen. Die klare Erkenntnis von Ursache und Wirkung und die daraus abgeleitete Entwicklung notwendiger Substanzen, die zielgerichtet und wirksam alles Leid verhindern, scheint in greifbare Nähe gerückt zu sein, der Traum von idealen Arzneimitteln.
Wo solche Errungenschaften gegeben sind, überwiegen jedoch nicht nur die positiven Aspekte. Scharlatanerie, Betrug und Missbrauch sind dort nicht weit, wo man Vorteile in finanzieller Sicht oder auch mit Blick auf Macht erringen kann. Dies haben unsere Vorfahren im Mittelalter zur Genüge erfahren, wenn Händler mit billigen Tinkturen über Land zogen und diese als Wunderheilmittel anpriesen oder Quacksalber sich mit selbigen Zugang zu den höchsten Fürsten- und Königshäusern verschafften. Hatte man Glück, so war es nur Wasser. Im schlimmeren Fall – wie nicht selten vorgekommen – waren es verunreinigte oder sogar giftige Substanzen, die so manchem Gutgläubigen den Tod brachten, der eigentlich das ewige Leben erwartete.
Doch was heißt Mittelalter? Auch wenn man in unserer heutigen, hoch technologisierten Zeit nicht unbedingt mehr von Scharlatanerie sprechen mag, so ist der Handel mit jenen Glücksbringern, vielleicht etwas moderner, über die mittlerweile sehr offenen Ländergrenzen und Kontinente hinweg nicht immer von der gewünschten notwendigen Seriosität geprägt. Und die Etikettierung von Billigprodukten als Markenartikel darf auch heute noch als Betrug bezeichnet werden, genauso wie der Konsum von Drogen unter die Kategorie Missbrauch fällt. Es sind aber nicht immer nur jene absichtlich betrügerischen und missbräuchlichen Vorgehen, die in diesem Umfeld Sorgen bereiten. Das komplexe und nicht ganz simple Thema der Arzneimittelherstellung birgt darüber hinaus eine Fülle an nicht immer frühzeitig erkennbaren Gefahren für den späteren Verbraucher. Ob es nun jene unglücklicherweise immer wieder im Betriebsalltag vorkommenden menschlichen oder technischen Fehler sind, die zu physikalischen, chemischen oder mikrobiellen Verunreinigungen führen oder ob eine neue Verfahrensvariante im Rahmen der Prozessoptimierung zu einer ungewollt negativen Veränderung im Reinheitsprofil führt – all dies sind Risiken. Zusammen mit jenen Risiken, die aus unter Umständen nicht vollständig erforschten Wirkungsweisen eines Medikaments resultieren, lassen sie den Ruf nach mehr Schutz des Verbrauchers laut werden, den Ruf nach Arzneimittelsicherheit.
Das Thema „Arzneimittelsicherheit“ ist komplex und lässt sich sicher nicht nur auf eine einzelne Maßnahme reduzieren. Ganz im Gegenteil, heute nimmt dieses Thema einen sehr breiten Raum ein und eine Vielzahl von Institutionen und Einrichtungen beschäftigt sich ausgiebig damit, Regeln, Überwachungs- und Kontrollmaßnahmen sowie alle erforderlichen Hilfsmittel bereitzustellen, die dazu beitragen sollen, dass Arzneimittel mit dem Höchstmaß an Sorgfalt und Sicherheit entwickelt und hergestellt werden, um somit den notwendigen Verbraucherschutz gewährleisten zu können. Anhand von Abb. 1.1 soll auf diese heute bestehenden Maßnahmen und Instrumentarien kurz eingegangen werden.
Der Entwicklungsweg eines Arzneimittels ist mittlerweile geprägt von einer Entwicklungszeit, die nicht selten deutlich über 10 Jahren liegt und durch Entwicklungskosten, die sich zunehmend der Grenze von 1 Mrd. Euro nähern. Eine überdimensionale Wandtafel im Deutschen Museum in München [1] stellt diese Entwicklung eindrucksvoll dar. Dabei findet der erste Schritt im Rahmen der Wirkstoffsuchforschung noch immer überwiegend im Labor statt, auch wenn die moderne Computertechnologie mit Molecular Modelling bereits ganz andere Wege aufzeigt. Im Labor werden, die Entwicklung der ersten Verfahrensschritte eingeschlossen, zunächst alle notwendigen Daten zur Substanz sowie zu den möglichen Synthese- und Herstellungswegen erarbeitet und dokumentiert. Es wird die Datenbasis für das spätere Arzneimittel und dessen Herstellung geschaffen. Dabei ist einerseits zu unterscheiden zwischen der Herstellung des reinen Wirkstoffs (der eigentlich wirksame Bestandteil eines Arzneimittels) und andererseits der Entwicklung der geeigneten Darreichungsform (z. B. Tablette, Kapsel oder Spritze), bei der Wirk- und Hilfsstoff (z. B. Wasser, Glucose, Gelatine etc.) in der sogenannten galenischen Entwicklung zum anwendungsfertigen Endprodukt formuliert, d. h. zusammengebracht werden.
Abb. 1.1 Maßnahmen zur Arzneimittelsicherheit.
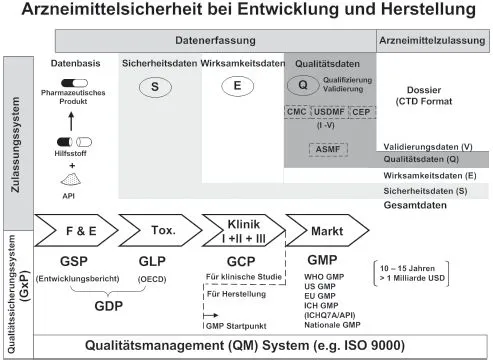
Liegen erste Erfolg versprechende Ergebnisse vor, so werden diese im nächsten Schritt im Rahmen der toxikologischen Studien weitergehend, jetzt schwerpunktmäßig mit Blick auf mögliche, von der Substanz ausgehende Gefahren, abgesichert. Reagenz-, aber auch Tierversuche werden herangezogen, um nun den die Sicherheit des Arzneimittels betreffenden Datenpool im Rahmen der sogenannten präklinischen Studien zu schaffen.
Ist auch diese Hürde genommen, so folgt der langwierigste und auch teuerste Abschnitt in der Entwicklung eines Arzneimittels, die klinischen Studien. In insgesamt drei in diesem Bereich formal ausgewiesenen Untersuchungsphasen (Phase I bis III) werden noch einmal die sicherheitskritischen Aspekte in der Anwendung, nun aber direkt am Menschen, ausgetestet [2, 3]. Es werden Untersuchungen zur geeigneten Dosierung, zur Wirkungsweise, zu Unverträglichkeiten und ggf. Nebenwirkungen sowohl an einzelnen Probanden als auch an einem immer größer werdenden Versuchskollektiv ausgetestet. Die Wirksamkeit, der eigentliche Zweck des Arzneimittels, steht jetzt unter Abwägung von Nutzen und Risiko im Mittelpunkt der Betrachtungen. Es gibt auch noch eine Phase IV, die unter dem Begriff der Pharmacovigilance [4] läuft. Hierbei handelt es sich aber um eine Langzeitverifizierung des sich bereits auf dem Markt befindlichen Produkts, die in dem hier dargestellten Entwicklungspfad noch keine Rolle spielt.
Parallel zur klinischen Prüfung laufen die verfahrenstechnischen Entwicklungen auf Hochtouren. Es gilt den geeigneten und – jetzt auch wirtschaftlich – optimalen Herstellungsweg zu finden. Ausgehend von ersten Versuchen im Labor führt der Weg oft über kleinere Pilotanlagen und über Technikumsentwicklungen schließlich in die für die Vermarktung vorgesehene Produktionsanlage. Dabei ist es wichtig, dass mit Beginn der klinischen Studien, spätestens aber ab Phase II, das Verfahren schon weitgehend entwickelt ist und für die Probanden Produkte eingesetzt werden, die mit der späteren Marktware vergleichbar bzw. identisch sind. Gegebenenfalls sind zusätzliche, ein mögliches Scale-up betreffende Nachweise zu erbringen, die belegen, dass das Produkt aus den klinischen Prüfungen die gleichen Qualitäten und Eigenschaften zeigt wie jenes Produkt, das in der endgültigen Produktionsanlage hergestellt wird (s. Abschnitt 3.2.1).
In Bezug auf die von dem Arzneimittel eventuell ausgehenden Risiken und die zum Schutz des Verbrauchers zu ergreifenden Maßnahmen sind zwei wesentliche Aktionsfelder zu unterscheiden: die Arzneimittelzulassung, in deren Rahmen heute sehr intensiv und nach sehr strikten und strengen Regeln geprüft wird, ob ein Arzneimittel überhaupt auf den Markt verbracht werden darf und die Implementierung von Qualitätssicherungssystemen, nach denen auf den unterschiedlichen Stufen gearbeitet werden muss und die teilweise der behördlichen Überwachung unterliegen, um sicherzustellen, dass über die Prozesse die notwendige Qualität gewährleistet ist.
Die Arzneimittelzulassung beruht im Wesentlichen auf Daten, die auf den unterschiedlichsten Entwicklungsstufen, die ein Arzneimittel durchläuft, systematisch gesammelt und ausgewertet werden (s. oberer Teil in Abb. 1.1), schließt aber am Ende auch Ergebnisse aus Audits ein, im Rahmen derer das jeweils implementierte Qualitätssicherungssystem überprüft und beurteilt wird. Die Daten stammen aus der Phase der Entwicklung, der toxikologischen und der klinischen Studien sowie aus der endgültigen Produktion. Man spricht von den Basisdaten (body of data), den sicherheitsrelevanten Daten (safety data), den die Wirksamkeit betreffenden Daten (efficacy data) und den Qualitätsdaten (quality data), die am Ende noch um die Validierungsdaten ergänzt werden. All diese Daten müssen säuberlichst zusammengetragen und nach Vorgaben, die in nationalen und internationalen Regelwerken [5, 6] ausführlich beschrieben sind, in einem Zulassungsdossier erfasst und beurteilt werden. Dabei ist nach einem festen Format und einer fest vorgegebenen Gliederung vorzugehen, die unter dem Begriff „Common Technical Document“ (CTD-Format) zusammengefasst sind [7]. Auch die Prüfung des Dossiers durch Behörden und Fachexpertengremien ist sehr detailliert in Regelwerken vorgegeben, soll hier aber nicht weiter vertieft werden. Der für Wirkstoffe hinsichtlich Zulassung wichtige und interessante Teil befindet sich dabei im Abschnitt des sogenannten Modul 3 gemäß ICH-Leitfaden M4, in dem die für die Herstellung und Qualitätssicherung wichtigen Daten und Informationen untergebracht sind. Diese Informationen zum Zulassungsdossier können dabei auf unterschiedlichste Art und Weise mit entsprechenden Vor- und Nachteilen vom Wirkstoffentwickler bzw. -hersteller an die Behörde bzw. den End-arzneimittelhersteller weitergegeben werden. Die bekanntesten Varianten sind der Drug Master File (DMF, heute in Europa ASMF = Active Substance Master File) und das CEP (Certification of Suitability to the Monographs of the European Pharmacopoeia) [8].
Was jedoch nutzen die einmalig gesammelten, bewerteten und aufgezeichneten Daten, wenn im Rahmen der Herstellung durch menschliches oder technisches Versagen Fehler auftreten, die die Qualität des Arzneimittels im Nachhinein negativ beeinflussen? Oder wenn gar die für das Zulassungsverfahren erarbeiteten Daten nicht verlässlich und aussagekräftig sind, weil nicht nach entsprechenden Standards gearbeitet und dokumentiert wurde? Die gewünschte Sicherheit und Zuverlässigkeit lässt sich also nur erreichen, wenn zusätzlich zu den etablierten Zulassungsabläufen auch Verfahren und Vorgehensweisen fixiert werden, welche sowohl im Bereich der Entwicklung als auch bei der späteren Produktion die dauerhafte Qualität der Erzeugnisse sicherstellen. Diese üblicherweise in Anweisungen (Verfahrens- oder Arbeitsanweisungen) zusammengefassten und festgeschriebenen Standards stellen das als Pendant zur Zulassung geforderte Qualitätssicherungssystem dar (s. unterer Teil der Abb. 1.1), wobei die hierzu zumeist offiziell erlassenen Vorgaben heute weltweit als „Good-Practices“-Regeln bekannt, etabliert und in weiten Teilen behördlich gefordert sind. Im Labor- bzw. Entwicklungsbereich spricht man allgemein von den „Good Science Practices“ (GSP) bzw. den „Good Laboratory Practices“ (GLP), wobei beiden die Forderung nach „Good Documentation Practices“, d. h. die Forderung nach einer guten, ausführlichen, aussagekräftigen und zuverlässigen Dokumentation gemein ist. Die klinischen Studien unterliegen den „Good-Clinical-Practices“ (GCP)-Regeln, während die eigentliche Herstellung sowohl des Produkts, welches in der Klinik Anwendung findet, als auch des fertigen Marktprodukts den „Good-Manufacturing-Practices“ (GMP)-Regeln folgen muss. Die „Good-Practices“-Regeln sind dabei stets der sehr weit gefasste und offen formulierte, auf die Produktqualität ausgerichtete Rahmen, der von den Anwendern in spezifische Regeln umgesetzt werden muss und der in das gesamtgültige Qualitätsmanagementsystem des jeweiligen Unternehmens eingebettet sein sollte.
Man erkennt, das Thema der Arzneimittelsicherheit ist so komplex und umfassend, dass es in einem Werk allein sicher nie oder nur sehr oberflächlich beschrieben werden kann. Die nachfolgenden Kapitel konzentrieren sich daher auch allein auf das Thema „GMP“, d. h. auf jenes für die Herstellung relevante Qualitätssicherungssystem und dort auch nur auf ein spezielles Unterthema, die Qualifizierung und Validierung, welches eine Hauptforderung aus den GMP-Regeln darstellt. Im Brennpunkt stehen dabei Anlagen zur Herstellung von Wirkstoffen, also die pharmazeutisch aktiven Bestandteile eines Arzneimittels.
Zur Übersicht: In Kapitel 2 wird zunächst auf den Begriff GMP, seine Historie und Bedeutung eingegangen. Das Kapitel 3 vermittelt die wesentlichen Grundlagen zu dem zentralen Thema „Validierung“, die jeder kennen sollte, wenn er sich eingehender mit der Thematik beschäftigen will. In Kapitel 4, dem zentralen Kapitel, werden dann detaillierte Informationen und Empfehlungen zur Umsetzung gegeben, wobei im Wesentlichen von einem „Musterkonzept“ ausgegangen wird, welches sich über Jahre hinweg in zahlreichen Projekten bewährt hat. Es erhebt nicht den Anspruch, in jedem Punkt optimal und für jeden Fall geeignet zu sein und dass ihm vonseiten des Lesers zwingend gefolgt werden muss, wenn dieser bereits bessere oder vergleichbare Lösungen für seinen Anwendungsfall hat. Dennoch können den Ausführungen sicher die einen oder anderen wertvollen Anregungen entnommen werden. Kapitel 5 behandelt das spannende Thema der integrierten Anlagenqualifizierung bzw. die Fragestellung, wie planende und bauende Ingenieure mit Qualifizierungsingenieuren so optimiert Hand in Hand arbeiten können, dass die notwendigen Qualifizierungsmaßnahmen mit einem Minimum an Zeit- und Kostenaufwand erledigt werden können. Kapitel 6 greift das Thema der externen Vergabe von Qualifizierungs- bzw. Validierungsleistungen auf und gibt Tipps, was dabei beachtet werden sollte. Kapitel 7 beschäftigt sich mit dem Erhalt des validierten bzw. qualifizierten Zustands und der sich daraus ableitenden Daueraufgabe, während das letzte Kapitel 8 die für viele sicher interessante Frage aufwirft, ob die Qualifizierung und Validierung den Ruf nach einem neuen Berufsbild laut werden lässt.
Dem Leser seien viel Spaß und nützliche Anregungen gewünscht.
2
GMP-Grundlagen
2.1 Der Begriff GMP
Good Manufacturing Practice – oder kurz GMP – ist ein Schlagwort, dem heute kaum jemand entgehen kann, wenn er im pharmazeutischen Umfeld tätig ist, ob als Hersteller, als Zulieferer oder Dienstleister. Die Regeln der Guten Herstellungspraxis – wie es im Deutschen heißt – sind ein auf die Produktqualität ausgerichtetes Qualitätssicherungssystem, das darauf abzielt, Produkte stets in gleichbleibender und zuverlässiger Qualität herzustellen und damit letztendlich den Anwender, in den meisten Fällen den Patienten, vor unerwünschten Nebenwirkungen zu schützen.
Eine „Ganze Menge Probleme“ oder eine „Ganze Menge Papier“ (engl.: „Give me more paper“) sind andere vielfach zu findende Übersetzungen für die Abkürzung GMP. Sie machen deutlich, dass es sich hierbei wohl um ein Thema handelt, das nicht gänzlich unumstritten und auch nicht ohne Fragen ist. Gerade Anfang der Neunzigerjahre hatte man GMP auch sehr eng mit den DIN ISO 9000 Qualitätsnormen in Verbindung gebracht und das Ganze als Steigerung dieser ohnehin schon sehr umstrittenen Standards angesehen. Als deutlich überzogene Forderungen und als reine Geschäftemacherei wurden die GMP-Regeln teilweise in der Presse dargestellt.
In der Tat, so alt das Thema auch sein mag (erste Grundregeln wurden 1968 von der WHO eingeführt), birgt es noch immer viele Unsicherheiten, gerade auch dann, wenn es um die inhaltliche Umsetzung geht. Wer konkret muss denn eigentlich nach GMP-Regeln arbeiten? Gilt dies schon im Labor oder auch bei sehr frühen Prozessstufen? Wie weit reicht GMP in der Prozesskette zurück? Welche Regelwerke gibt es und wie verbindlich sind diese? Was steht konkret in diesen Regeln? Was muss ich tun oder beachten und wie setze ich GMP-Anforderungen konkret um? Wie sieht eine GMP-gerechte Produktionsanlage aus? Was ist an Kosten und zusätzlichem Personalaufwand zu erwarten und habe ich überhaupt eine Chance, bezogen auf meine Produkte, all diese Anforderungen wirtschaftlich vernünftig umzusetzen?
Diese oder ähnliche Fragen stellen sich heute immer noch sehr viele Betriebe, die mit dem Thema zu tun haben. Insbesondere die Hersteller von Wirkstoffen, d. h. die Chemische Industrie, haben das grundlegende Problem, sehr häufig mit großen Mengen an Niedrigpreis-Produkten im Vergleich zu den Hochpreis-Produkten der pharmazeutischen Industrie auf den Markt gehen zu müssen. Solche Chemieprodukte tragen oft nur schwer diese zusätzlichen Aufwände und Kosten. Erschwerend kommt hinzu, dass in der chemischen Industrie oftmals keine reinen GMP-Betriebe, sondern Mischungen von GMP- und Nicht-GMP-Betrieben vorkommen. Und zu guter Letzt machen die zunehmend steigende Anzahl an Kunden- und Behördenaudits und die damit verbundenen Forderungen die Situation für die Verantwortlichen auch nicht gerade einfacher (Abb.2.1 ).
Heute lassen sich die Probleme mit GMP speziell im Wirkstoffbereich auf drei wesentliche Kernpunkte zusammenfassen:
1. Nicht in allen Fällen ist es klar und eindeutig, ob GMP-Regeln aus gesetzlicher Sicht eingehalten werden müssen oder nicht. Während es für Fertigarzneimittelhersteller hier keine Diskussionen gibt, da der Gesetzgeber dies klar und eindeutig für alle Phasen des Prozesses zwingend vorgeschrieben hat, hängt es bei den Herstellern von aktiven pharmazeutischen Bestandteilen (engl.: API = Active Pharmaceutical Ingredient = Wirkstoff) und Herstellern von Hilfsstoffen (engl.: excipient) von unterschiedlichsten Faktoren ab, wie z. B. der Verfahrensstufe, auf der sich das hergestellte Produkt befindet, der Entwicklungsstufe, auf der sich das Verfahren befindet und dem für das Produkt vorgesehenen Markt, d. h. vom vorgesehenen Exportland.
2. GMP-Regelwerke, -Richtlinien, ergänzende -Leitfäden, -Standards und -Empfehlungen existieren mittlerweile in einer nahezu unüberschaubaren Fülle, sodass kaum noch jemand wirklich in der Lage ist, all diese Regelwerke im Detail zu kennen und zu beherrschen. Jedes Jahr geben Behörden und Industrieverbände eine Vielzahl neuer Entwürfe und Diskussionsgrundlagen heraus, stets mit dem Ziel, schon bestehende GMP-Regeln um weitergehende Interpretationen zu ergänzen. Die Flut an Regularien, Richtlinien und Standards wächst kontinuierlich an.
Abb. 2.1 Probleme rund um GMP.
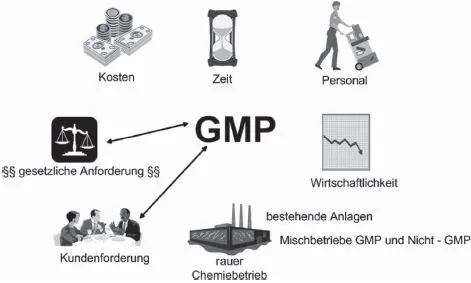
3. Selbst das intensive Lesen und Studieren all dieser Regeln, Leitlinien und Dokumente führt oft nicht zum gewünschten Ergebnis, d. h. diejenige Lösung zu finden, nach der man konkret sucht. Zu unterschiedlich sind die Produkte, zu verschieden die Prozesse und Verfahren und die vorgegebenen Randbedingungen, als dass es wirklich möglich wäre, Regeln derart zu gestalten, dass all diese Aspekte im Detail je berücksichtigt werden könnten. Demzufolge sind und bleiben GMP-Regelwerke und -Richtlinien stets sehr allgemein, ohne wirklich ins Detail zu gehen und spezifische Probleme zu beschreiben. Der große Interpretationsfreiraum ist das Markenzeichen der GMP-Regeln und kann auf der einen Seite als ein herausragender Vorteil gesehen werden, da er die notwendige Flexibilität bietet, stellt aber gleichzeitig auf der anderen Seite für den nach einer konkreten Lösung Suchenden oft ein unüberwindbares Hindernis dar. Fluch und Segen liegen hier dicht beieinander und lassen zentrale Fragen offen:
– Wo beginnt GMP?
– Welche Regelwerke gibt es?
– Wie verbindlich sind die Regelwerke?
– Wie setze ich die Anforderungen konkret um?
Umso wichtiger ist es, zum einen die Anforderungen und die Bedeutung von GMP in vollem Umfang und richtig zu erfassen und zu interpretieren und zum anderen pragmatische Lösungswege zu finden, um auf der einen Seite nicht zu viel, auf der anderen Seite aber auch nicht zu wenig für die Umsetzung zu tun. Was heißt nun GMP konkret und welche Anforderungen verstecken sich dahinter?
Good Manufacturing Practice (GMP) (dt.: Gute Herstellungspraxis) ist ein Begriff, der erstmals 1962 von der US-amerikanischen Überwachungsbehörde Food and Drug Administration (FDA) eingeführt wurde. Er steht synonym für eine Sammlung von Verhaltensmaßnahmen und Vorschriften, die bei der Herstellung und beim Umgang mit bestimmten Produkten (z. B. Arzneimittelprodukte) beachtet und eingehalten werden müssen. Eine erste offizielle von der EG 1989 herausgebrachte und noch heute gültige Definition besagt: „GMP ist der Teil der Qualitätssicherung, der gewährleistet, dass Produkte gleichbleibend nach den Qualitätsstandards produziert und geprüft werden, di...
Inhaltsverzeichnis
- Cover
- Title page
- Series
- Copyright
- Geleitwort Storhas
- Geleitwort Behrendt
- Vorwort
- Abknsverzeichnis
- 1: Einführung
- 2: GMP-Grundlagen
- 3: Grundlagen der Validierung
- 4: Validierungs-„How-to-do”
- 5: Integrierte Anlagenqualifizierung
- 6: Outsourcing von Validierungsaktivitäten
- 7: Change Control
- 8: Der Validierungsingenieur als neuer Beruf
- 9: Literatur
- 10: Verzeichnisse und Anlagen
- Index
Häufig gestellte Fragen
Ja, du kannst dein Abo jederzeit über den Tab Abo in deinen Kontoeinstellungen auf der Perlego-Website kündigen. Dein Abo bleibt bis zum Ende deines aktuellen Abrechnungszeitraums aktiv. Erfahre, wie du dein Abo kündigen kannst
Nein, Bücher können nicht als externe Dateien, z. B. PDFs, zur Verwendung außerhalb von Perlego heruntergeladen werden. Du kannst jedoch Bücher in der Perlego-App herunterladen, um sie offline auf deinem Smartphone oder Tablet zu lesen. Erfahre, wie du Bücher herunterladen kannst, um sie offline zu lesen
Wir sind ein Online-Lehrbuch-Abo, bei dem du für weniger als den Preis eines einzelnen Buches pro Monat Zugang zu einer ganzen Online-Bibliothek erhältst. Mit über 1 Million Büchern zu über 990 verschiedenen Themen haben wir bestimmt alles, was du brauchst! Erfahre mehr über unsere Mission
Achte auf das Symbol zum Vorlesen bei deinem nächsten Buch, um zu sehen, ob du es dir auch anhören kannst. Bei diesem Tool wird dir Text laut vorgelesen, wobei der Text beim Vorlesen auch grafisch hervorgehoben wird. Du kannst das Vorlesen jederzeit anhalten, beschleunigen und verlangsamen. Erfahre mehr über die Funktion „Vorlesen“
Ja! Du kannst die Perlego-App sowohl auf iOS- als auch auf Android-Geräten nutzen, damit du jederzeit und überall lesen kannst – sogar offline. Perfekt für den Weg zur Arbeit oder wenn du unterwegs bist.
Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Ja, du hast Zugang zu GMP-Qualifizierung und Validierung von Wirkstoffanlagen von Ralf Gengenbach im PDF- und/oder ePub-Format sowie zu anderen beliebten Büchern aus Technik & Maschinenbau & Chemie- & Biochemietechnik. Aus unserem Katalog stehen dir über 1 Million Bücher zur Verfügung.