Wie in kaum einem anderen Bereich der Medizin hat die molekulargenetische Forschung das Wissen um Muskelerkrankungen vermehrt. Erste spezifische Therapieoptionen sind bereits verfügbar. Dieses Buch ermöglicht einen raschen, an den Erfordernissen der Patientenversorgung orientierten Überblick zu Diagnose und Therapie der verschiedenen Muskelerkrankungen und schließt damit eine Lücke im deutschsprachigen Raum. Geschrieben aus der Praxis für die Praxis folgt es einem einheitlichen didaktischen Konzept. Tabellen, Diagramme, Fallbeispiele und Praxishinweise erleichtern dem Leser den Zugang.

eBook - ePub
Neuromuskuläre Erkrankungen
- 332 Seiten
- German
- ePUB (handyfreundlich)
- Über iOS und Android verfügbar
eBook - ePub
Neuromuskuläre Erkrankungen
Über dieses Buch
375,005 Studierende vertrauen auf uns
Zugang zu über 1 Million Titeln zu einem fairen monatlichen Preis.
Mit unseren Lerntools kannst du noch effizienter lernen.
Information
1 Muskelerkrankungen: Einteilung und Symptome
1.1 Einleitung
Im Zeitalter von Stroke units, DRG-Abrechnung, Behandlungspfaden und dergleichen besteht die Gefahr, dass die Muskelerkrankungen immer weniger klinisch-neurologisch beachtet werden. Da diese Erkrankungen zudem selten sind, wird es in der fachärztlichen Ausbildung und in der weiteren ärztlichen Arbeit immer schwieriger, ausreichend eigene Erfahrungen zu dieser Krankheitsgruppe zu erwerben, um den betroffenen Patienten und deren Familien gerecht zu werden. Die Einstellung mancher Neurologen, es sei müßig, sich mit solch „unbehandelbaren Raritäten“ abzugeben, hat es schon immer gegeben. Diese Position ist jedoch aus mehreren Gründen falsch:
- Die korrekte Diagnose und die sich daraus ergebende Prognose des weiteren Krankheitsverlaufs ist Voraussetzung für die Beratung von Muskelkranken, an der sie ihre weitere Lebensplanung orientieren können. Erfahrungsgemäß sind viele Muskelkranke sehr an einer möglichst genauen Aufdeckung des bei ihnen vorliegenden Krankheitsprozesses interessiert. Entsprechend willigen sie auch in invasive diagnostische Verfahren ein, wie in eine Muskelbiopsie, obwohl sie wissen, dass sich daraus für sie zumeist keine kausal angreifende Therapieoption ergibt. Die korrekte Diagnose ist weiterhin essenziell für eine genetische Beratung der Patienten und ihrer Familien bei Vorliegen einer hereditären Myopathie.
- Effiziente Therapiemöglichkeiten existieren für einige der in diesem Buch vorgestellten Krankheitsbilder, wie z. B. für die Myasthenie. Unter den monogenetisch verursachten Muskelerkrankungen ist beim M. Pompe mit der Enzymersatztherapie eine erste kausal wirksame Therapie verfügbar. Der rasante Fortschritt der molekulargenetischen Forschung lässt hoffen, dass in naher Zukunft auch kausal-therapeutische Interventionen für andere hereditäre Myopathien verfügbar werden.
- Es gibt inzwischen ein breites Spektrum symptomatischer Therapien zur Verbesserung der Lebenssituation bei neuromuskulären Erkrankungen. Vielfach kann damit auch bei schwerer Behinderung eine subjektiv hohe Lebensqualität erreicht werden. Ein Beispiel ist die nächtliche nicht-invasive Beatmung zur Behandlung einer chronisch-respiratorischen Insuffizienz bei Muskelschwäche, die die Leistungsfähigkeit und das Befinden während der Tagestunden erheblich bessern kann. Entscheidend für symptomatische Therapiemaßnahmen ist dabei die korrekte Indikationsstellung zum richtigen Zeitpunkt im Rahmen einer interdisziplinären Zusammenarbeit. Auch geht es darum, schwer wiegende Komplikationen rechtzeitig zu erkennen, wie z. B. Herzrhythmusstörungen bei der Emery-Dreifuss-Muskeldystrophie.
- Wie bei allen chronischen Erkrankungen hat der Arzt nicht nur die Rolle des „Medikamentenverordners“. Er sollte vielmehr den Patienten unterstützend begleiten und ihm mit menschlichem Verständnis ärztlich helfen.
Beide Autoren betreuen seit vielen Jahren Patienten mit neuromuskulären Erkrankungen. Wir verstehen dieses Buch daher auch als Erfahrungsbericht für jüngere Kollegen. Der Umfang erlaubt keineswegs eine erschöpfende Darstellung des Themas. Es gibt allerdings einen Einstieg in Diagnose und Therapie muskulärer Erkrankungen und bietet eine Orientierungshilfe bei der Interpretation und Beurteilung der Befunde. Wir hoffen, dass unser Buch dazu beiträgt, die diagnostische und therapeutische Versorgung von Muskelpatienten zu verbessern. Keineswegs sind Muskelerkrankungen so selten, wie es für den nur wenig Informierten den Anschein haben mag.
1.2 Klassifikation neuromuskulärer Erkrankungen
Neuromuskuläre Erkrankungen sind Erkrankungen von Komponenten der motorischen Einheit. Diese ist definiert als die Funktionseinheit von spinalem oder bulbären Motoneuron, dessen Zellkörper im Vorderhorn des Rückenmarks oder im Hirnstamm lokalisiert ist, dem dazugehörigen Axon, das über Vorderwurzel und peripheren Nerven zum Muskel gelangt, sich dort aufzweigt und über die neuromuskuläre Endplatte Kontakt aufnimmt zu einer unterschiedlich großen Anzahl von Muskelfasern in einem quergestreiften Muskel. Der Inhalt des Buches ist entsprechend der in Abbildung 1.1 vorgenommenen Klassifikation gegliedert, beginnend mit den Erkrankungen im Muskel.
Muskeldystrophien
Muskeldystrophien sind hereditäre Erkrankungen der Muskulatur, bei denen es im Verlauf des Lebens zu einem zunehmenden Untergang von Muskelfasern und fettig-fibrösem Ersatz quergestreifter Muskelzellen kommt. Dabei gibt es eine große Bandbreite klinischer Schweregrade. Allen Dystrophien gemeinsam ist der fortschreitende Untergang von Muskelzellen und die langsame Zunahme der klinischen Symptome im Verlauf des Lebens.
Seit der 1987 erfolgten Klonierung des Dystrophin-Gens und der daraus resultierenden genetischen Klärung der Duchenne- und der Becker-Muskeldystrophie hat sich die nosologische Einteilung der Muskeldystrophien erheblich verändert. Scheinbar festgefügte klinische Entitäten haben sich in genetisch eigenständige Unterformen aufgelöst. Es hat sich aber auch die große klinische Varianz einzelner genetischer Defekte gezeigt.
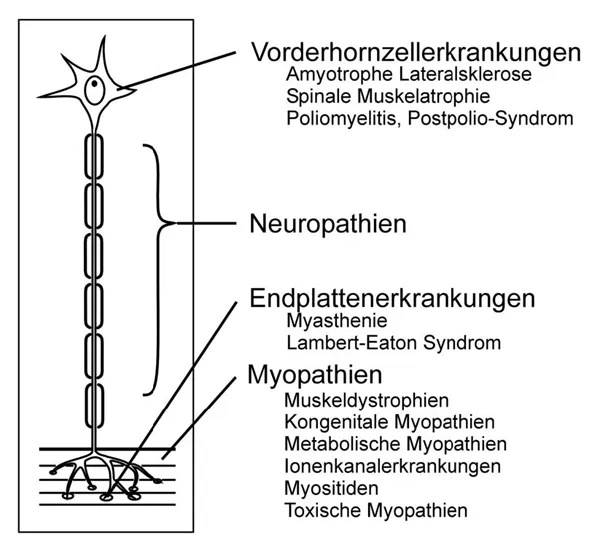
Abb. 1.1: Klassifikation neuromuskulärer Erkrankungen
So war die Diagnose Gliedergürteldystrophie (engl. limb girdle muscular dystrophy, LGMD) über Jahrzehnte eine Ausschlussdiagnose innerhalb des Spektrums neuromuskulärer Erkrankungen, die sich vorwiegend im Schulter- und/oder Beckengürtel manifestieren. Ende der 80er Jahre wurde diese Entität sogar als obsolet angesehen, da man davon ausging, dass letztlich alle Fälle eines Gliedergürtelsyndroms sich einer der damals bekannten anderen neuromuskulären Erkrankung zuordnen lassen würden (Jerusalem und Sieb 1992). Heute werden sechs autosomal-dominant vererbte (LGMD1A bis LGMD1F) und 15 autosomal-rezessiv vererbte Gliedergürteldystrophien (LGMD2A bis LGMD2O) unterschieden. Diesen liegt ein breites Spektrum genetischer Defekte zugrunde, wobei einige Unterformen nach wie vor molekular ungeklärt sind.
Kongenitale Myopathien
Kongenitale Myopathien sind ebenfalls vererbte Muskelerkrankungen. Sie sind gekennzeichnet durch einen Beginn in der frühen Kindheit, oft schon im Neugeborenenalter und durch einen eher statischen Krankheitsverlauf. Die Kinder zeigen eine Hypotonie, eine Trinkschwäche und entwickeln Skelettdeformitäten wie eine Skoliose. Bei manchen kongenitalen Myopathien kommt es im Laufe der Entwicklung sogar zu einer ganz allmählichen Besserung. Die Mehrzahl der kongenitalen Myopathien zeigt charakteristische morphologische Veränderungen der Muskelhistologie. Daneben gibt es auch kongenitale Muskeldystrophien, bei denen der Muskel dystroph verändert ist, wobei der klinische Verlauf in den frühen Lebensjahren jedoch eher den kongenitalen Myopathien ähnelt.
Metabolische Myopathien
Den metabolischen Myopathien liegt eine Störung des Energiestoffwechsels der Muskelzelle zugrunde. Obwohl es sich um erbliche Erkrankungen handelt, manifestieren sie sich jedoch vielfach erst im jüngeren Erwachsenenalter. Zu den metabolischen Myopathien gehören die mitochondrialen Myopathien mit Defekten der Atmungskette, Störungen des Lipidstoffwechsels, die Glykogenosen mit Enzymdefekten des Glukose- und Glykogenstoffwechsels sowie Störungen des Adenin-Nukleotid-Stoffwechsels. Ein Teil dieser Stoffwechseldefekte führt zu einer Muskelschwäche, wie z. B. der M. Pompe (Glykogenspeichererkrankung wegen eines Mangels an saurer alpha-Glukosidase), andere zeigen eine Belastungsintoleranz bis hin zu einem akutem Muskelzellzerfall, der Rhabdomyolyse, die durch Myoglobinurie (dunkle Verfärbung des Urins) in schweren Fällen zum akuten Nierenversagen führen kann.
Ionenkanalerkrankungen
Hierzu gehören die Myotonien und die periodischen Paralysen, die nicht zu einem zunehmenden Muskelzellverlust führen. Bei den myotonen Dystrophien handelt es sich um Multi-Systemerkrankungen mit einem besonderen RNAvermittelten Pathomechanismus (vgl. Kapitel 6.4.1).
Myositiden
Myositiden sind erworbene Autoimmunerkrankungen des Muskels, deren rechtzeitige Diagnose vor allem wegen der besonderen therapeutischen Konsequenzen von hoher Dringlichkeit ist. Jedoch ist bei der Einschlusskörpermyositis (inclusion body myositis) – der häufigsten erworbenen Muskelerkrankung jenseits des 50. Lebensjahrs – eine wirksame Therapie nicht bekannt. Die Muskelhistologie zeigt bei der Einschlusskörpermyositis neben entzündlichen Zellinfiltraten auch zytoplasmatische Einschlüsse u. a. mit Nachweis von phosphorylierten Neurofilamenten. Die Pathogenese der Einschlusskörperchenmyositis ist ungeklärt.
Toxische Myopathien
Toxische Myopathien werden verursacht durch exogen zugeführte Substanzen. Dies sind häufig Medikamente, z. B. Zidovudin als anti-retrovirale Substanz und Cholesterinsenker. Alkohol ist ebenfalls myotoxisch. Endokrine Störungen können auch zu einer Muskelaffektion führen. Eine wichtige iatrogene Myopathie ist die Steroidmyopathie durch eine hochdosierte Glukokortikoiddauertherapie. Andere Ursachen für eine Muskelschädigung sind beispielsweise schwere Elektrolytstörungen wie eine Hypokaliämie oder eine Ischämie.
Endplattenerkrankungen
Bei den Endplattenerkrankungen liegt eine Störung der Signalübertragung zwischen Nerv und Muskel vor. Häufigstes Beispiel ist die Myasthenia gravis als Autoimmunerkrankung. Es gibt jedoch auch kongenitale Myasthenie-Syndrome und toxisch bedingte Störungen der Endplattenfunktion wie den Botulismus.
Vorderhornzellerkrankungen
Bei den Vorderhornzellerkrankungen kommt es zu Muskelschwäche und Muskelschwund wegen eines einmaligen (z. B. bei der Poliomyelitis) oder zunehmenden Motoneuronverlustes im Rückenmark und Hirnstamm. Die zahlenmäßig größte Gruppe stellt die sporadische amyotrophe Lateralsklerose dar, es gibt jedoch auch zahlreiche hereditäre Motoneuronerkrankungen.
1.3 Muskuläre Symptome
1.3.1 Schwäche
Das Kardinalsymptom von Muskelerkrankungen ist eine muskuläre Schwäche. Bei im Kindesalter beginnenden Erkrankungen führt dies zu einer verzögerten motorischen Entwicklung. Die motorischen Meilensteine der kindlichen Entwicklung werden nicht altersgemäß erreicht. Die körperliche Leistungsfähigkeit bleibt unter dem Level gesunder Altersgenossen, wie sich spätestens beim Schulsport zeigt. Andere, auch hereditäre Myopathien, manifestieren sich jedoch erst im Erwachsenenalter mit einer progredienten Muskelschwäche. Die Frage nach Alltagsaktivitäten, also z. B. nach der maximalen Gehstrecke, Treppensteigen, Aufrichten aus der Hocke, Körperpflege, Nahrungsaufnahme und anderem, hilft, den Grad der Behinderung einzuschätzen.
Häufig fällt als erstes ein ungewöhnliches Gangbild auf. Manchmal werden Alltagsverrichtungen wie das Aufrichten aus der Hocke zunehmend erschwert und schließlich unmöglich. Eine Schwäche des M. quadriceps wird zunächst deutlicher beim Treppenabsteigen registriert als beim Hinaufsteigen. Bei einer Schwäche des Musculus gluteus medius kommt es zu einem typischen wiegenden Gangbild (Trendelenburg-Zeichen) – in der Standbeinphase kann das Becken nicht mehr horizontal gehalten werden und kippt verstärkt nach unten. Bei ausgeprägteren Paresen muss die kontralaterale Schwungbeinhüfte mit einer Rumpfdrehung nach vorne gebracht werden, was zu einem entenähnlichen „Watschelgang“ führt. Bei ausgeprägter Beckengürtelschwäche, wie sie z. B. bei der Duchenne-Muskeldystrophie auftritt, gelingt das Aufstehen vom Boden nur im Vierfüßlerstand und durch ein Hochklettern mit den Armen an sich selbst („Gowers-Manöver“, s. Kapitel 5).
Bei Prüfung der Muskelkraft zeigt sich häufig ein Gliedergürtelsyndrom, d. h. eine proximal betonte, symmetrische Schwäche der Extremitäten ohne Sensibilitätsstörungen. Seltener kommen bei Muskelerkrankungen auch distale, mimische, okuläre oder oropharyngeale Schwerpunkttypen vor. Distal verteilte Extremitätenparesen lassen zunächst eher an neurogene Erkrankungen denken (symmetrisch bei Neuropathien, asymmetrisch bei Motoneuronerkrankungen). Eine distale Betonung der Paresen findet man bei Muskelerkrankungen am häufigsten bei der myotonen Dystrophie, aber auch bei einer Reihe seltener Muskelerkrankungen wie der z. B. distalen Myopathien und myofibrillären Myopathien. Bei der Einschlusskörpermyositis sind neben dem M. quadriceps insbesondere auch die Fingerbeuger betroffen und bei dem Slow channel-Syndrom, einem seltenen kongenitalen Myasthenie-Syndrom die Hand- und Fingerstrecker.
- Eine Facies myopathica ist gekennzeichnet durch ein ausdruckloses Gesicht mit einer Schwäche der mimischen Muskulatur, die sich u. a. in einem signe des cils (Sichtbarbleiben der Wimpern bei Lidschluss) äußert. Durch die bei kongenitalen Myopathien früh einsetzende Schwäche der orofazialen Muskulatur kommt es zur Ausbildung eines hohen (gotischen) Gaumens.
- Typisches Erstsymptom der Myasthenia gravis sind Doppelbilder durch eine Schwäche der äußeren Augenmuskeln. Bei bestimmten Muskelerkrankungen ist auch die Ausbildung einer externen Op...
Inhaltsverzeichnis
- Deckblatt
- Titelseite
- Impressum
- Inhaltsverzeichnis
- Vorwort
- 1 Muskelerkrankungen: Einteilung und Symptome
- 2 Diagnostik und Therapie von Muskelerkrankungen
- 3 Muskeldystrophien
- 4 Kongenitale Myopathien
- 5 Metabolische Myopathien
- 6 Myotonien, periodische Paralysen und andere muskuläre Ionenkanalerkrankungen
- 7 Entzündliche Muskelerkrankungen
- 8 Toxisch und endokrin bedingte Myopathien
- 9 Myasthenia gravis und andere Endplattenerkrankungen
- 10 Motorische Systemerkrankungen
- 11 Schlussbetrachtung
- Abkürzungen
- Literatur
- Stichwortverzeichnis
Häufig gestellte Fragen
Ja, du kannst dein Abo jederzeit über den Tab Abo in deinen Kontoeinstellungen auf der Perlego-Website kündigen. Dein Abo bleibt bis zum Ende deines aktuellen Abrechnungszeitraums aktiv. Erfahre, wie du dein Abo kündigen kannst
Nein, Bücher können nicht als externe Dateien, z. B. PDFs, zur Verwendung außerhalb von Perlego heruntergeladen werden. Du kannst jedoch Bücher in der Perlego-App herunterladen, um sie offline auf deinem Smartphone oder Tablet zu lesen. Erfahre, wie du Bücher herunterladen kannst, um sie offline zu lesen
Perlego bietet zwei Abopläne an: Elementar und Erweitert
- Elementar ist ideal für Lernende und Profis, die sich mit einer Vielzahl von Themen beschäftigen möchten. Erhalte Zugang zur Basic-Bibliothek mit über 800.000 vertrauenswürdigen Titeln und Bestsellern in den Bereichen Wirtschaft, persönliche Weiterentwicklung und Geisteswissenschaften. Enthält unbegrenzte Lesezeit und die Standardstimme für die Funktion „Vorlesen“.
- Pro: Perfekt für fortgeschrittene Lernende und Forscher, die einen vollständigen, uneingeschränkten Zugang benötigen. Schalte über 1,4 Millionen Bücher zu Hunderten von Themen frei, darunter akademische und hochspezialisierte Titel. Das Pro-Abo umfasst auch erweiterte Funktionen wie Premium-Vorlesen und den Recherche-Assistenten.
Wir sind ein Online-Lehrbuch-Abo, bei dem du für weniger als den Preis eines einzelnen Buches pro Monat Zugang zu einer ganzen Online-Bibliothek erhältst. Mit über 1 Million Büchern zu über 990 verschiedenen Themen haben wir bestimmt alles, was du brauchst! Erfahre mehr über unsere Mission
Achte auf das Symbol zum Vorlesen bei deinem nächsten Buch, um zu sehen, ob du es dir auch anhören kannst. Bei diesem Tool wird dir Text laut vorgelesen, wobei der Text beim Vorlesen auch grafisch hervorgehoben wird. Du kannst das Vorlesen jederzeit anhalten, beschleunigen und verlangsamen. Erfahre mehr über die Funktion „Vorlesen“
Ja! Du kannst die Perlego-App sowohl auf iOS- als auch auf Android-Geräten nutzen, damit du jederzeit und überall lesen kannst – sogar offline. Perfekt für den Weg zur Arbeit oder wenn du unterwegs bist.
Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Ja, du hast Zugang zu Neuromuskuläre Erkrankungen von Jörn P. Sieb,Bertold Schrank, Thomas Brandt, Reinhard Hohlfeld, Johannes Noth, Heinz Reichmann, Thomas Brandt,Reinhard Hohlfeld,Johannes Noth,Heinz Reichmann im PDF- und/oder ePub-Format sowie zu anderen beliebten Büchern aus Medicine & Neurology. Aus unserem Katalog stehen dir über 1 Million Bücher zur Verfügung.