Die Amyotrophe Lateralsklerose (ALS) ist die häufigste Motoneuron-Erkrankung bei Erwachsenen (ca. 6000 Patienten in Deutschland). Die ALS führt zu fortschreitender Muskellähmung verbunden mit Muskelsteifheit und Bewegungsunfähigkeit bei meist klarem Verstand. Der Tod tritt in der Regel 3-5 Jahre nach Krankheitsbeginn ein. Die im Verlauf auftretenden Beschwerden und die psychosoziale Begleitung der Patienten und ihrer Familien sind eine Herausforderung für das Betreuungsteam. Dieses praxisorientierte Werk bietet konkrete Hinweise für eine ganzheitliche Palliativbetreuung bei ALS. In jedem Abschnitt werden konkrete Problemlösungsstrategien beschrieben, angepasst an den deutschen Versorgungskontext.
Häufig gestellte Fragen
Gehe einfach zum Kontobereich in den Einstellungen und klicke auf „Abo kündigen“ – ganz einfach. Nachdem du gekündigt hast, bleibt deine Mitgliedschaft für den verbleibenden Abozeitraum, den du bereits bezahlt hast, aktiv. Mehr Informationen hier.
Derzeit stehen all unsere auf Mobilgeräte reagierenden ePub-Bücher zum Download über die App zur Verfügung. Die meisten unserer PDFs stehen ebenfalls zum Download bereit; wir arbeiten daran, auch die übrigen PDFs zum Download anzubieten, bei denen dies aktuell noch nicht möglich ist. Weitere Informationen hier.
Mit beiden Aboplänen erhältst du vollen Zugang zur Bibliothek und allen Funktionen von Perlego. Die einzigen Unterschiede bestehen im Preis und dem Abozeitraum: Mit dem Jahresabo sparst du auf 12 Monate gerechnet im Vergleich zum Monatsabo rund 30 %.
Wir sind ein Online-Abodienst für Lehrbücher, bei dem du für weniger als den Preis eines einzelnen Buches pro Monat Zugang zu einer ganzen Online-Bibliothek erhältst. Mit über 1 Million Büchern zu über 1.000 verschiedenen Themen haben wir bestimmt alles, was du brauchst! Weitere Informationen hier.
Achte auf das Symbol zum Vorlesen in deinem nächsten Buch, um zu sehen, ob du es dir auch anhören kannst. Bei diesem Tool wird dir Text laut vorgelesen, wobei der Text beim Vorlesen auch grafisch hervorgehoben wird. Du kannst das Vorlesen jederzeit anhalten, beschleunigen und verlangsamen. Weitere Informationen hier.
Ja, du hast Zugang zu Palliative Care bei Amyotropher Lateralsklerose von Johanna Anneser, Gian Domenico Borasio, Wendy Johnston, David Oliver, Andrea Winkler, Sibylle Tönjes, Gian Domenico Borasio, Monika Führer, Maria Wasner, Ralf J. Jox im PDF- und/oder ePub-Format sowie zu anderen beliebten Büchern aus Medizin & Neurologie. Aus unserem Katalog stehen dir über 1 Million Bücher zur Verfügung.
In diesem Kapitel sollen folgende Fragen beantwortet werden: Was ist die amyotrophe Lateralsklerose (ALS)? Was bedeutet im Unterschied hierzu die Diagnose einer Motoneuronerkrankung? Wir beginnen mit einer Darstellung des typischen klinischen Bildes der ALS und der bei dieser Erkrankung notwendigen Diagnostik und gehen kurz auch auf andere Krankheiten ein, die zur Degeneration von Motoneuronen führen oder mit ähnlichen Symptomen einhergehen. Anschließend befassen wir uns mit der Krankheitsentstehung und -entwicklung der ALS und geben einen Überblick über die Forschungsergebnisse auf den Gebieten der Molekulargenetik und Zellbiologie. Zum Schluss gehen wir auf Therapien ein, die den Krankheitsverlauf beeinflussen können.
Was ist eine Motoneuronerkrankung?
Der Begriff Motoneuronerkrankung umfasst mehrere unterschiedliche Krankheitsbilder, bei denen sich der Krankheitsprozess vornehmlich an den motorischen Nervenzellen (Motoeurone) abspielt. Klinische, histopathologische und molekulargenetische Studien haben dazu beigetragen, viele dieser Krankheiten von der typischen ALS abzugrenzen. Die Amyotrophe Lateralsklerose wird in vielen Ländern auch als »typische« Motoneuronerkrankung bezeichnet. Im englischsprachigen Raum ist mit »motor neuron disease« daher oft die ALS gemeint (und nicht die ganze Gruppe der Krankheiten, die die Motoneurone betreffen). In den Vereinigten Staaten von Amerika ist sie auch bekannt als Lou Gehrig’s disease – nach dem berühmten, an ALS verstorbenen Football-Spieler.
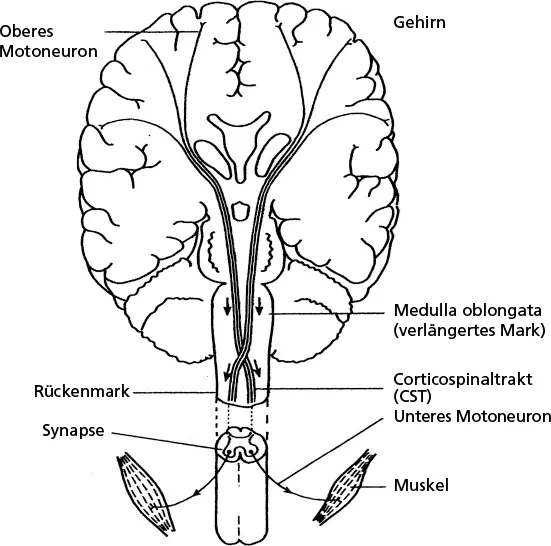
Ursprünglich ging man davon aus, dass es sich um eine Erkrankung der Muskulatur handelt, bis Charcot im Jahr 1869 klinisch-pathologische Studien veröffentlichte, in denen er korrekt die Degeneration von motorischen Neuronen als Ursache des Muskelschwunds identifizierte (Charcot JM, Joffroy A 1869). Diese Krankheit unterscheidet sich von anderen Krankheiten des Bewegungsapparates durch eine kombinierte Degeneration des 1. und 2. Motoneurons (MN) (die Nervenleitungsbahnen sind in
Abb. 1.1 dargestellt). Als andere Bezeichnung ist auch zentrales und peripheres Motoneuron gebräuchlich.
Abb. 1.1: Nervenbahnen in Rückenmark und Gehirn
Die Zellkörper der 2. Motoneurone (2.MNs) finden sich in Rückenmark und Hirnstamm und haben über die peripheren Nerven direkten Kontakt mit Muskelfasern, die sie aktivieren. Wenn die 2. MNs degenerieren, kommt es zu einer Schwäche der von ihnen innervierten Muskeln. Ein weiteres Symptom hierfür sind die Muskelzuckungen (Faszikulationen) und Muskelschwund (Atrophie). Die 1. Motoneurone (ZMNs) liegen im sog. Motorischen Kortex, im Stirnlappen des Großhirns. Die Fortsätze (Axone) dieser Neurone verlaufen im Rückenmark nach unten zu den 2. MNs, die durch sie aktiviert werden. Wenn die 1. MNs degenerieren, entwickelt sich eine Muskelsteifigkeit (Spastik) der betroffenen Muskeln und es kommt zu gesteigerten Muskeleigenreflexen. Als weiteres Zeichen findet sich bisweilenein sog. »Babinski-Zeichen«: die Großzehe wird beim Bestreichen der Fußsohle nach oben gezogen statt sich nach unten einzurolllen). Bei den meisten ALS-Patienten finden sich kombinierte Zeichen der 1. MNs und 2. MNs. Liegen nur Zeichen der 2. MNs vor, wird die Krankheit als progressive Muskelatrophie (PMA) bezeichnet, bei ausschließlichen 1. MN-Symptomen als primäre Lateralsklerose (PLS). Histopathologische Studien legen nahe, dass diese Syndrome jeweils Unterformen der ALS sind und sich an den Enden eines gemeinsamen klinischen Spektrums befinden. Bei der Progressiven Muskelatrophie überwiegt demnach eine Schädigung der 2. Motoneurone und bei der primären Lateralsklerose die der 1. Motoneurone. Es ist interessant, dass diese beiden Varianten langsamer progredient verlaufen und mit einem längeren Überleben verbunden sind als eine »klassische« ALS.
Die Diagnose einer ALS wird klinisch gestellt und durch technische Zusatzuntersuchungen gestützt. In der Regel geht die ALS mit progressiver Schwäche und Atrophie der Extremitätenmuskulatur und der zum Sprechen und dem Schlucken notwendigen Muskulatur, einher. Die Motoneurone, die die Augenbewegungen sowie die Schließmuskeln von Harnblase und Darm kontrollieren, bleiben typischerweise ausgespart. Das Gleiche gilt für die sensiblen Neurone, die z. B. Schmerz oder Berührung übermitteln und die Neurone des vegetativen Nervensystems. Die diagnostischen Kriterien der ALS wurden bei einer Konferenz in dem spanischen Schloss El Escorial festgelegt und 2002 aktualisiert (»El Escorial Kriterien«) (
Abb. 1.2). Sie werden als Einschlusskriterien für Therapiestudien verwendet und verdeutlichen, dass für die Diagnose einer sicheren ALS -Symptome des 1. und 2. MNs in mehreren Bereichen der Muskulatur nachgewiesen werden müssen.
Es gibt zahlreiche Motoneuronerkrankungen, die – zumindest im Anfangsstadium – mit einer ALS verwechselt werden können. Aufgrund der Bedeutung einer ALS-Diagnose für den Patienten muss diese möglichst zweifelsfrei gestellt und alle anderen in Betracht kommenden Erkrankungen ausgeschlossen werden. Die häufigste Differenzialdiagnose ist eine Kompression des Rückenmarks und/oder einer Nervenwurzel durch eine Degeneration der Wirbel und der Bandscheiben. Hierdurch kann es in einer oder mehreren Extremitäten zur schmerzlosen Muskelatrophie, zu Muskelschwäche und Faszikulationen kommen. Daher wird bei Verdacht auf ALS in der Regel eine Magnetresonanztomografie (MRT) des Rückenmarks und der Nervenwurzeln durchgeführt. Die MRT deckt auch in seltenen Fällen vorkommende Rückenmarkstumoren oder eine Syringomyelie (zystische Erweiterung im Rückenmark) auf. Eine weitere Krankheit, die ausgeschlossen werden muss, ist die multifokale motorische Neuropathie (MMN). Bei dieser Autoimmunerkrankung greifen Autoantikörper selektiv die motorischen Nerven an und es kommt zu einer -in der Regel- asymmetrischen Schwäche der oberen Extremitäten. Bei der MMN ist die Nervenleitgeschwindigkeit der motorischen Nerven oft an mehreren Stellen reduziert – dies kann im Einzelfall jedoch nur schwer nachzuweisen sein. Außerdem finden sich im Serum Anti-GM1-Gangliosid-Antikörper (Pestronk A et al. 1988). Zeichen einer Schädigung des 1. MNs finden sich bei dieser Erkrankung nicht. Die MMN hat eine deutlich bessere Prognose und kann sich durch eine immunsuppressive Behandlung z. B. mit intravenös oder subkutan verabreichtem humanem Immunglobulin bessern.
Die Kennedy-Krankheit oder spinobulbäre Muskelatrophie (SBMA) ist zwar insgesamt selten, wird aber häufig falsch als ALS diagnostiziert. Dieses Syndrom betrifft ausschließlich die 2. Motoneurone und tritt nur bei erwachsenen Männern auf. Es kommt zur progressiven Muskelatrophie insbesondere der Zunge und zum Haltetremor der Hände. Weitere Symptome sind eine Hodenatrophie und eine Gynäkomastie (Vergrößerung der Brustdrüse) durch niedrige Androgenspiegel (Harding AE et al. 1982). Bei der Messung der Nervenleitgeschwindigkeit findet sich oft eine leichte sensible Neuropathie, obwohl die klinische Untersuchung häufig keine Sensibilitätsverluste zeigt. Ursache ist die Expansion einer CAG-Nukleotidrepeat-Sequenz in dem für den Androgenrezeptor kodierenden Gen auf dem X-Chromosom (La Spada AR et al. 1991). Das CAG-Triplet kodiert für die Aminosäure Glutamin. Durch die Expansion ändert sich die Konformation des Androgenrezeptorproteins: Diese akkumulieren in den Motoneuronen und gewinnen hierdurch zellschädigende Eigenschaften.
Noch seltener ist die autosomal-rezessive spinale Muskelatrophie (SMA). Sie manifestiert sich meistens im Säuglingsalter oder in der Kindheit, gelegentlich aber auch als SMA Typ IV erst im Erwachsenenalter. Dieses langsam progrediente reine 2. MN-Syndrom geht mit Muskelatrophie, Muskelschwäche und fehlenden Muskeleigenreflexen einher (Dubowitz V 1995). In mehr als 95% der SMA-Fälle ist das sog. Survival-Motor-Neuron-Gen (SMN-Gen) deletiert (Lefebvre S et al. 1995).
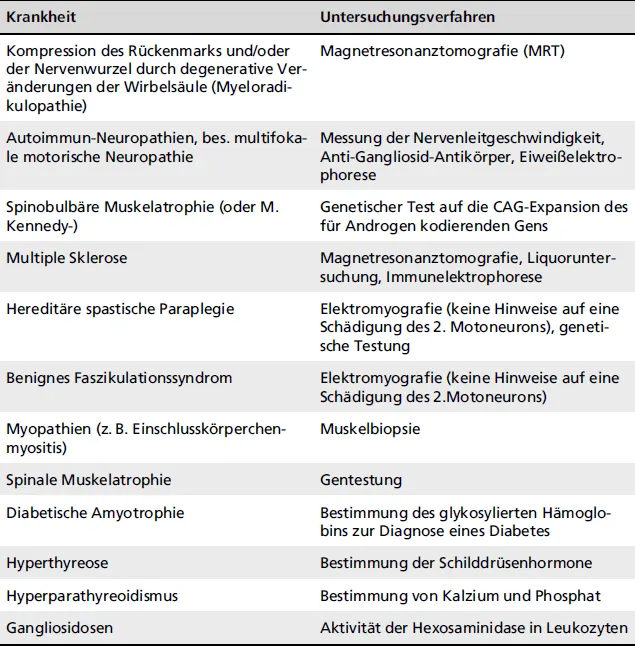
Zur Differenzialdiagnose gehören noch zahlreiche weitere Krankheiten, wie die Multiple Sklerose und die hereditäre spastische Paraplegie, die zu Symptomen der 1. MNs führen. Bei Muskelzuckungen ohne fokale Muskelatrophie besteht der Verdacht auf ein benignes Faszikulationssyndrom oder auf Erkrankungen der Schilddrüse und der Nebenschilddrüsen, das durch Bestimmung der Hormonspiegel diagnostiziert werden kann (
Tab. 1.1).
Tab. 1.1: Differenzialdiagnosen der amyotrophen Lateralsklerose
KrankheitUntersuchungsverfahren
Klinischer Verlauf der ALS
Bei den meisten ALS-Patienten (85%) beginnt die Muskelschwäche schleichend an einer Extremität, typischerweise mit einer Schwäche beim Greifen oder einer Fußheberschwäche. Da in der Folge weitere periphere Motoneurone im Rückenmark zugrunde gehen, kommt es zu Faszikulationen und einer fortschreitenden Muskelatrophie der betroffenen Extremität. Die Spastik – als Zeichen einer Beteiligung des 1. MNs – kann der Muskelschwäche aber auch vorausgehen oder gleichzeitig mit dieser auftreten. In einem fortschreitenden Prozess zeigen auch andere Extremitäten Lähmungen, Faszikulationen und Atrophie, sodass bei vielen Patienten schließlich alle Gliedmaßen betroffen sind. Seltener beginnen die Beschwerden im Bereich der Sprech-, Kau- und Schluckmuskulatur (»Bulbärbereich«) (15%) mit verwaschener Sprache oder Schluckstörungen (Haverkamp LJ et al. 1995). Da diese Symptome durch Degeneration der 2. MNs im Hirnstamm (früher als »Bulbus« bezeichnet) verursacht sind, wird a diese Verlaufsform oft als progressive Bulbärparalyse bezeichnet (
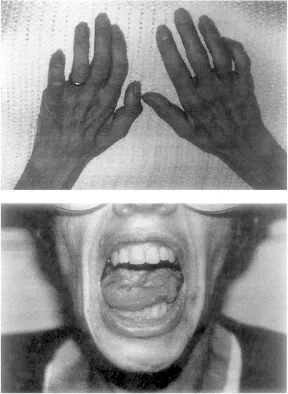
Abb. 1.3). Gelegentlich finden sich bulbäre Symptome auch als Folge einer Schädigung des 1. MNs: Es kommt zu einer Spastik der Zunge und zu einer Erhöhung des Muskeltonus im Kiefer. Hier kann es auch zu ruckartigen Bewegungen (Kloni) des Unterkiefers kommen.
Abb. 1.3: Klinisches Bild der amyotrophen Lateralsklerose. Deutlich zu erkennen ist die Muskelatrophie an Händen und Zunge.
Dieser Symptomkomplex wird oft als Pseudobulbärparalyse bezeichnet. Bulbärparalyse und Pseudobulbärparalyse können in einer ALS münden: Es kommen bei diesen Patienten im Verlauf Symptome der Extremitätenmuskulatur hinzu und eine typische ALS entwickelt sich. Umgekehrt treten bei etwa 90% der Patienten, bei denen die ALS an den Extremitäten beginnt, schließlich bulbäre Symptome auf; nur bei sehr wenigen Patienten und überwiegend Männern wird der Hirnstamm überhaupt nicht in die Erkrankung miteinbezogen.
Die ALS führt meist frühzeitig zu Behinderungen und schreitet – wenn auch in unterschiedlichem Tempo – unaufhaltsam voran. Die meisten Patienten können in einem fortgeschrittenen Stadium nicht mehr eigenständig gehen, essen, sprechen und schlucken und sich auch nicht mehr eigenständig um ihre Körperhygiene kümmern. Viele Menschen empfinden es als besonders grausam, dass die intellektuellen Fähigkeiten überwiegend erhalten bleiben. Die Patienten sind sich ihres Zustandes bewusst, jedoch vollständig gelähmt und durch die Einschränkungen im sozialen Kontakt und der Kommunikation häufig isoliert. Bei etwa 10% der Patienten treten deutliche Persönlichkeits-, Verhaltens- und Sprachstörungen im Sinne einer frontotemporalen Demenz auf. Bis zu 30–40% der Patienten weisen ähnliche, aber mildere kognitive Defizite auf (
Kap. 8).
Die meisten Patienten versterben durch eine Schwäche der Atemmuskulatur. Diese wird oft lange unterschätzt, da die Mobilität der Patienten eingeschränkt ist und ein Abfall der Vitalkapazität auf 60% des Normwertes zunächst häufig asymptomatisch bleibt. Die mittlere Überlebenszeit nach dem Auftreten der ersten Symptome beträgt 3 Jahre. Nur 25% der Patienten überleben 5 Jahre und 10% sind nach 10 Jahren noch am Leben (Kondo K 1995). Bei älteren Frauen mit initialer Bulbärsymptomatik ist die Prognose deutlich schlechter, bei der langsam progredienten ALS junger Männer mit primärer Extremitätenschwäche besser.
Wie wird die Diagnose ALS gestellt?
Die ALS ist eher selten und die Diagnose kann zu Beginn der Erkrankung schwierig sein. Die meisten Allgemeinmediziner werden im Laufe ihres Lebens höchstens einmal mit der ALS konfrontiert. Daher werden die Frühzeichen und -symptome oft übersehen und die Patienten erst zu Rheumatologen, Orthopäden oder Hals-Nasen-Ohren-Ärzten geschickt, bevor sie schließlich einem Neurologen vorgestellt werden. Die Symptome und die Befunde der körperlichen Untersuchung liefern zu Beginn oft nur Hinweise auf eine ALS, einen einfachen »Test auf ALS« gibt es nicht. Aufgrund der doch erheblichen Auswirkungen für den Patienten sprechen die Neurologen meist die Möglichkeit einer ALS erst an, wenn sie absolut sicher sind, dass die Diagnose zutrifft. Alle diese Faktoren tragen dazu bei, dass die Diagnose nach dem erstmaligen Auftreten der Symptome mit erheblicher Verzögerung gestellt wird.
Hilfreich sind alle Untersuchun...
Inhaltsverzeichnis
Deckblatt
Titelseite
Impressum
Inhalt
Vorwort
Vorwort zur deutschen Auflage
Abkürzungsverzeichnis
1 Die Amyotrophe Lateralsklerose (ALS)
2 Palliative Care
3 Kommunikation und Aufklärung
4 Entscheidungen treffen
5 Patientenverfügung und Advance Care Planning
6 Respiratorische Komplikationen
7 Dysphagie
8 Kognitive Dysfunktion
9 Schmerzen, psychischer Distress und andere Symptome
10 Psychosoziale Betreuung
11 Spititual Care
12 Physiotherapie
13 Ergotherapie
14 Logopädie
15 Pflege
16 Komplementär- und Alternativmedizin
17 End of Life-Care: ethische Aspekte
18 End of Life-Care bei ALS
19 Trauer
20 Keine Zeit zu verlieren: die Reise einer Familie von der Diagnose bis zur Trauerphase