![]()
Chapter 1
Introduction
1.1 The beginnings - Some history
In 1929, in a paper entitled “Quantum Mechanics of Many-Electron Systems”, the physicist P. A. M. Dirac made the following provocative statement:1
“The underlying physical laws necessary for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known, and the difficulty is only that the exact application of these laws leads to equations much too difficult to be soluble. It therefore becomes desirable that approximate practical methods of applying quantum mechanics should be developed, which can lead to an explanation of the main features of complex atomic systems without too much computation.”
Presumably, the physicists who read this paper were mollified to some extent because some of their number would still be employed, but it seems that the paper was (fortunately) ignored by chemists, who carried on as usual. Indeed, in the eighty two years that have followed the appearance of Dirac’s paper, it is likely that more new chemical compounds have been recorded than in all the previous years. What also happened was the “approximate practical methods”, envisaged by Dirac, appeared, because of something that he only hinted at: “computation”. Dirac shared the Nobel Prize for Physics in 1933 with Erwin Schrödinger.
While the majority of chemists were doing their own things (“Stamp Collecting” according to Ernest Rutherford - another physicist, who ironically got a Nobel Prize for Chemistry!) some physical chemists and chemical physicists did take Dirac’s words to heart, and so ‘Theoretical Chemistry’ and then (when computers were invented) ‘Computational Chemistry’ came about.
1.2 About the book
This book is about “Molecular Modelling”. We have given it a subtitle “Computational Chemistry Demystified” because we believe that modelling provides an easy entry point into computational chemistry, a branch of chemistry that has hitherto been misunderstood and undervalued, and thence into the rest of chemistry. While chemists have used many sorts of models for a long time, we concentrate our attention on computer based modelling because now this is the method of choice, and the necessary equipment is, if not already available, easily affordable.
We follow the present introductory chapter with one that deals with the choice of computer equipment. We do this because we believe that success in modelling and computation can be critically dependent on making the right choice. However, if you have already purchased your computer for other purposes, do not despair, what we have written may still help you in improving its performance, and to adapt it for molecular modelling.
While Chapter 2 deals primaily with the business of acquiring a computer suitable for Molecular Modelling, it also deals with wider issues of operating systems and handling more than one computer. If you already have a computer, we show how with very simple modifications (or none at all) it can do the job better. This chapter will also help those responsible for organizing a course in Molecular Modelling for the first time.
In the later chapters we show how to get the necessary chemical structural information, in forms that can be used by a computer. There is a vast amount of this that is freely available; free in the sense that it is easy to acquire, and also that it is in many cases available without payment.
Chapter 3 deals with principles of molecular mechanics. This is the easiest form of molecular modelling calculation to understand. The basic ideas about structure optimisation by minimizing the energy associated with a structure, that are at the heart of molecular mechanics, can be transferred to molecular orbital methods. We do not deal with these methods in detail, preferring to point you in the direction of the many excellent books that do. We do discuss the ways in which you may gain access to the programs that perform molecular orbital calculation.
Chapter 4 introduces the tools provided by INTERCHEM that allow you to build organic chemical structures of gradually increasing complexity. You are shown how to optimize the structures and then compare them with structures determined experimentally. The methods and examples should be handled equally well by other molecular modelling packages.
In Chapter 5 attention is turned to the larger structures of proteins and nucleic acids. There is a brief discussion of the salient features that distinguish these structures from those of smaller organics structures. We show how to gain access to the vast resources of the Protein Data Bank, and how to best display these structures to gain insight of their functions, and analyse them in various ways. The program PRESTO is introduced to allow the analysis and comparison of protein sequences.
Since stereochemistry and conformational analysis play important parts in molecular modelling, and indeed are underlying themes in our book, we offer brief summaries of these subjects in Chapter 6.
While most methods of computational chemistry deal with molecules in some sort of ideal (gaseous) state, and have difficulty dealing with molecules in solution, Chapter 7 deals with molecular modelling applied to the crystalline state. This is an area where progress is beginning to be made. The methods of molecular mechanics allow the many of the properties of crystals to be understood, and progress has been made in predicting the space group in which a chemical entity may crystallize.
In Chapter 8 we return to the basics of data handling. The information that is needed to define a structure, and the way that it is stored, has often leads to problems for people starting out in molecular modelling. It is not possible to discuss all the data formats that have been used, so we concentrate on those that have been mentioned in this book, or are commonly used elsewhere. We show those parts of a data file that are essential, and those that are not.
Perhaps the most compelling reason for writing this book comes from the use of molecular modelling in connection with Drug Design and Medicinal Chemistry, and this, and other related applications are dealt with in Chapters 9 and 10. The high cost of bringing a new drug to market has always made the companies involved turn to all available tools that might minimize this cost. Pharmaceutical firms have been held to ransom by the high prices of modelling software, and as our token contribution to the reduction of this cost, the Interprobe software is offered without charge to all under the GNU General Public License.
Two optical discs holding this software are provided with this book. You may copy and distribute the contents of these, and mount the software on any number of computers, provided you abide by the terms and spirit of the license.2 The discs also have complete manuals for the programs INTERCHEM and PRESTO.
1.2.1 Some Caveats
It is perhaps necessary to define what we mean by a model in the context of chemistry. This is best done by showing what a model is not. Almost every molecular modelling software package will have an option (usually the default option) to show sulfur atoms as yellow balls. This does not imply that if we could actually see the atoms they would be coloured yellow. At the atomic scale the idea of colour is not meaningful. If you think carefully about it, every diagrammatic representation of a chemical structure is, to some extent, a model. The crystallographer who has, with great skill, determined the structure of (say) a protein is looking at a model. He or she is perhaps justified in being sceptical of a modeller who attempts to imbue a modification of such a structure with the same veracity as the original.3 Provided that these limits of modelling are kept in mind, molecular modelling has a vital role to play in chemical research and education.
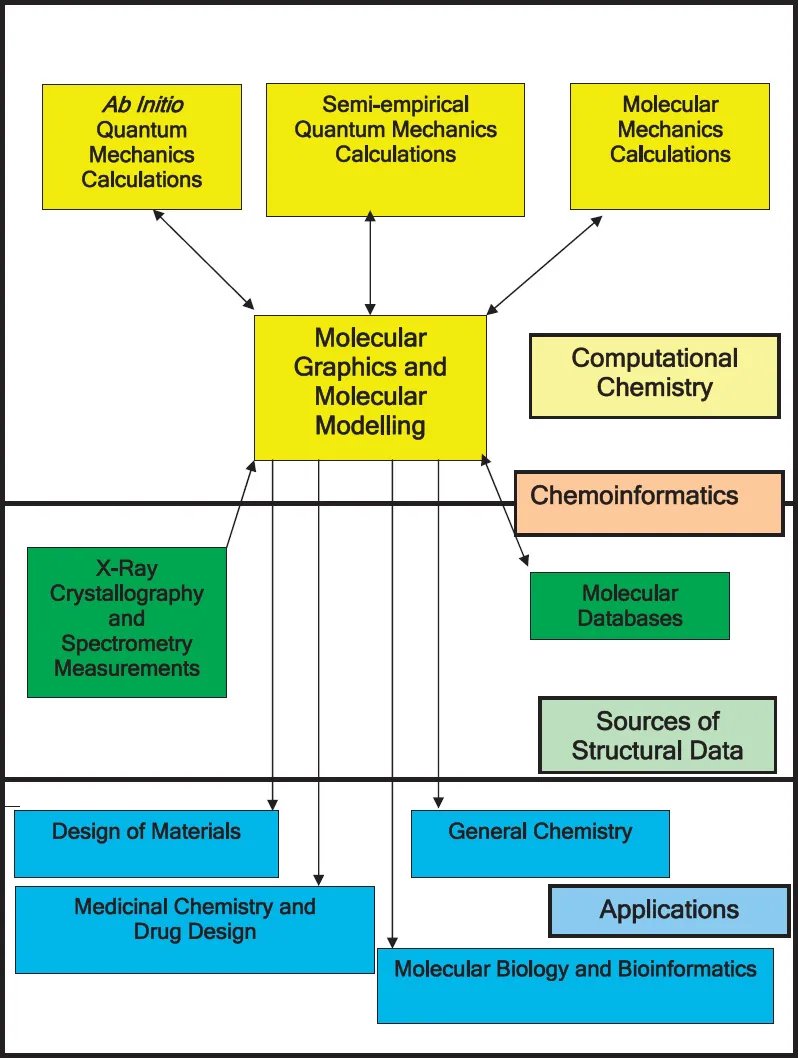
Molecular modelling is frequently dismissed as simply providing ‘pretty pictures’. But many prestigious scientific journals make a feature of artwork that can rely in one way or another on molecular modelling.4 To emphasize that modelling is respectable we would remind you, the reader, of the way that the structure of deoxyribonucleic acid was revealed by the extensive use of models.5 Figure 1.1 shows how molecular modelling underpins computational chemistry, and how some of the other topics covered in this book are related.
1.3 Getting started
The usual advice given to any one using a piece of equipment or software for the first time is ‘Read the Manual’. Experience has taught us that most people will ignore this. We acquiesce in this because we want to give you a taste of what molecular modelling can do. But we will assume that you have your computer set up, that the program INTERCHEM has been loaded on the machine, that you have started it by clicking on the appropriate icon on the desktop, and have pressed the escape key to get past the Welcome screen.6
Figure 1.1
Molecular Modelling and its Role in Chemistry
If you move the mouse sprite around the buttons that are at the bottom of the screen, relevant help sentences will appear that will explain what each button does. You can click on the buttons to gain access to various tools. Not all of these will be appropriate for our first example (the molecule valium); some are only relevant if you have two structures present; some are only relevant if you are viewing a large biomolecule or a crystal structure.
Using the mouse, click first on the button Get from Catalogue (Row 2/Column 8 in the Dual Display menu; henceforth we abbreviate the button coordinates to R2/C8), and then on buttons in the succession of menus that are revealed: Aromatics and Heterocyclics, Valium, Confirm. In the left hand window there will appear a simple ‘wire-frame’ structure of Valium (alias diazepam) (1).
What you have seen thus far is an entry point into molecular graphics, molecular modelling, and computational chemistry. In the remainder of this chapter, we will make use of INTERCHEM to explain the way these three subjects (and others) are connected, how you may gain entry to them, and profitably use them.
If you have succeeded so far, then you can load another structure into the right hand viewing area. Click on Structure B and repeat the steps used before but choose the structure Biotin (2).
The wire-frame depictions of valium and biotin show the bonds but gives no indication of the atoms that constitute the molecules. The identities of the atoms can be revealed in several ways; press each of the following (yellow) buttons in turn:
(1) Bali and Cylinder (R3/C9). This will show atoms as coloured balls (carbon and hydrogen - white, oxygen - red, nitrogen - blue, chlorine - green, sulfur - yellow), and the bonds as coloured cylinders (single - white, double - green, triple - blue, aromatic -cyan).
(2) CPK Atoms (R3/C10) This will show the atoms only as larger balls (approximately 1.5 times the van-der-Walls radii of the atoms).7
(3) Cylindrical Bonds (R4/C6). This will show the bonds only as coloured cylinders.
(4) Wire Frame (R3/...