![]()
Introduzione
La Fibrosi Cistica
Con circa 70.000 persone affette e 1000 nuovi casi l’anno, la Fibrosi Cistica (FC) è la malattia genetica letale più diffusa al mondo, con una mortalità che si attesta a circa il 90% dei casi [Misbahuddin M, 2017]. Un individuo su venticinque è portatore sano di questa patologia monogenica con ereditarietà autosomica recessiva, le cui manifestazioni cliniche sono causate da mutazioni del gene CFTR (Cystic Fibrosis Transmembrane Conductance Regulator).
L’omonima proteina, che da questo gene viene codificata, è un canale anionico regolato dall’cAMP, espresso sulla superficie apicale delle cellule epiteliali di differenti tessuti e organi secretori (polmoni, pancreas, intestino, genitali), dove consente il passaggio dello ione cloro, seguito per osmosi dal flusso di acqua, che rende quindi fluido il muco che riveste i suddetti tessuti.
Sono state identificate quasi duemila mutazioni del gene CFTR che determinano alterazioni strutturali e/o funzionali dell’omonimo canale e che causano quindi un ridotto trasporto dello ione cloro, predisponendo così, ad un ispessimento dello strato di secrezione mucosa con conseguente ostruzione e danno tissutale [Bradley S, 2016].
Le manifestazioni cliniche più frequenti di questa patologia multiorgano, sono infatti, ritardo nella crescita, causato da una ridotta secrezione di enzimi pancreatici e conseguente difficoltà nell’assorbimento di grassi e proteine, e alterazioni respiratorie che frequentemente evolvono in infezioni polmonari croniche [Castellani C, 2017].
Lo spesso strato di muco risulta essere infatti un ambiente ottimale per la proliferazione batterica, che determina uno stato di infiammazione cronica a cui segue un massivo intervento di neutrofili [Misbahuddin M, 2017].
Il meccanismo d’azione di questi leucociti, nel tentativo di ridurre l’infezione, causa tuttavia il rilascio di un’elevata concentrazione di acidi nucleici e proteine della matrice, che contribuiscono insieme al rilascio di elastasi e citochine proinfiammatorie, all’incremento della viscosità del muco e al danno tissutale [Misbahuddin M, 2017].
Le terapie per la FC, prima rivolte esclusivamente alla riduzione della sintomatologia, si stanno adesso sempre più rivolgendo alla causa fondante della patologia, nel tentativo di individuare una terapia univoca che sia in grado di ripristinare la funzionalità del canale CFTR e che sia valida per la grande varietà di mutazioni che colpiscono questo gene.
Il gene CFTR e le sue mutazioni
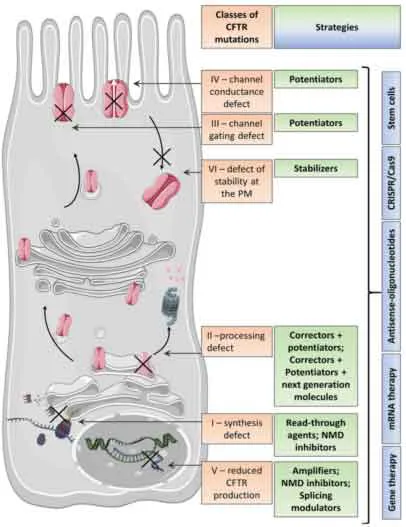
Il gene CFTR, lungo circa 230 kb, è localizzato sul braccio lungo del cromosoma 7 e codifica per una proteina di 1480 amminoacidi; al termine della traduzione, essendo una proteina di membrana, CFTR attraverserà reticolo endoplasmatico e apparato del Golgi, andando incontro a complesse modifiche post-traduzionali che le consentiranno di raggiungere il corretto ripiegamento. Le molteplici mutazioni a carico di questo gene possono essere distinte in sei classi in base all’alterazione causata e alla gravità delle manifestazioni cliniche [Iwona Pranke et al. 2019 ].
Figura. 1: C
lassificazione delle mutazioni a carico di CFTR e corrispondenti strategie terapeutiche [Iwona Pranke et al. 2019].
Classe I: riguarda il 2-5% della popolazione e comprende tutte le mutazioni che portano alla mancata sintesi della proteina e quindi alla sua totale assenza al livello della membrana [Misbahuddin M, 2017]. Un esempio sono le mutazioni nonsenso, che inserendo un codone di stop prematuro (PTC), generano un trascritto instabile, maggiormente soggetto alla degradazione da parte delle nucleasi e al decadimento mediato dal
pathway NMD (Nonsense Mediated Decay). Qualora questo mRNA riesca a raggiungere il ribosoma la proteina sintetizzata risulta essere tronca e quindi non funzionale [Iwona Pranke et al. 2019].
Classe II: di questa classe fa parte la mutazione più comune fra i soggetti affetti da FC, (presente nell’80% dei casi), ovvero la delezione della tripletta codificante per la fenilalanina in posizione 508 (F508del), a cui segue l’impossibilità di un corretto ripiegamento della proteina che la rende energicamente instabile e ne promuove quindi la degradazione con un meccanismo proteasoma dipendente. Come gli altri membri di questa classe, la F508del causa quindi un processamento anormale, determinando una proteina incapace di raggiungere la membrana plasmatica e quindi all’assenza del flusso di cloro [Bradley S Quon and Steven M Rowe, 2016].
Classe III: comprende mutazioni riguardanti la porzione regolativa del canale, causando quindi difetti nella fosforilazione e nella capacità dei domini NBDs di legare e/o idrolizzare l’ATP. La proteina riesce quindi ad inserirsi in membrana, ma, la mancata fosforilazione causa alterazioni nella regolazione del meccanismo di apertura del canale. Queste mutazioni vengono infatti definite mutazioni di
gating e determinano quindi una severa riduzione o totale assenza del flusso di cloro.
Classe IV: la quasi totalità delle mutazioni appartenenti a questa classe riguarda i domini formanti il poro di permeazione, causando così alterazioni nella permeabilità e una riduzione della probabilità di apertura del canale. Queste mutazioni portano ad una corretta sintesi della proteina che viene anche correttamente regolata, risultando quindi meno severe delle alterazioni appartenenti alle classi precedenti.
Classe V: le mutazioni appartenenti a questa classe riguardano regioni non codificanti del gene CFTR, come il promotore o sequenze introniche, con la possibilità di indurre l’incorporazione di esoni criptici o alterare il corretto splicing. La proteina risultante, di conseguenza, se pur correttamente funzionante, è presente in quantità ridotte rispetto ad una condizione fisiologica.
Classe VI: comprende mutazioni che causano una ridotta stabilità della proteina e un incremento del suo turnover; determinano quindi una riduzione del flusso di cloro attraverso la membrana apicale dei tessuti coinvolti.
Le differenti classi determinano manifestazioni fenotipiche ampiamente eterogenee sia nel grado di severità della patologia, sia nella velocità di progressione della stessa, con le forme più severe che si identificano nelle classi I, II e III.
Una maggiore variabilità è determinata anche dalle diverse combinazioni alleliche in cui le mutazioni possono trovarsi a livello genomico.
Riveste, quindi, sempre più importanza la genotipizzazione, non solo per conferma della diagnosi, ma anche per l’identificazione del trattamento più appropriato e la personalizzazione della terapia [Ramsey et al. 2011].
Le manifestazioni fenotipiche della patologia sono però significativamente differenti anche in pazienti presentanti le stesse mutazioni, con un decorso non sempre prevedibile sulla base degli esami clinici e molecolari. Sul fenotipo finale influiscono infatti fattori ambientali e variazioni geniche che riguardano locus diversi da quello di CFTR. Ne sono esempi, i polimorfismi a carico dei geni coinvolti nel pathway NMD o del gene codificante per il fattore di crescita trasformante β (TGFβ). [Drumm et al. 2005]. Per queste ragioni, l’identificazione di una terapia univoca è un obiettivo complesso da raggiungere. Nonostante ciò, alla terapia genica, vanno progressivamente aggiungendosi trattamenti con piccole molecole capaci di agire sul difetto proteico o al livello del trascritto di CFTR.
Struttura della proteina CFTR
CFTR è una proteina canale facente parte della superfamiglia delle ATP-binding cassette (ABC). Nonostante condivida con le proteine ABC le principali caratteristiche strutturali, La proteina CFTR possiede proprietà funzionali uniche che lo rendono essenziale per il mantenimento della fisiologia dei tessuti in cui è espresso.
Gli altri membri di questa superfamiglia sfruttano infatti l’idrolisi dell’ATP per determinare un trasporto attivo dei rispettivi substrati, al contrario di CFTR in cui il passaggio degli ioni avviene secondo gradiente elettrochimico e il legame dell’ATP è funzionale solo all’apertura del canale [Theodoulou and Kerr, 2015].
CFTR è un canale relativamente non selettivo, permeabile agli anioni monovalenti grazie alla presenza, sulla superficie del poro di permeazione, di residui di arginine e lisine, che, con la loro carica positiva, attraggono gli anioni e respingono i cationi [Fangyu Liu et al., 2017]. Il poro è definito da due domini transmembrana (TMD1, TMD2), ciascuno formato da 6 α-eliche che si connettono alla componente citoplasmatica del canale, composta da due domini NBD (nucleotide-binding domain), con il ruolo di legare l’ATP. Secondo il meccanismo di apertura del canale, l’idrolisi dell’ATP avverrà soltanto quando i due NBDs formeranno un dimero, che consentirà c...