![]()
Chapter 1
The Role of DNA Damage in Cancers Caused by Chemicals
Miriam C. Poirier
Abstract
Many cancers caused by chemical carcinogens do so through a mutagenic mechanism initiated by the formation of carcinogen–DNA damage. Some chemical carcinogens can damage DNA with no metabolic intervention, and others must be biotransformed. DNA adduct formation is considered necessary but not sufficient for tumorigenesis, as dosimetry for DNA adducts and tumorigenesis has been amply demonstrated in experimental models. Once methods became available to measure DNA adducts in humans, studies showed widespread distribution of DNA adducts in human tissues, dosimetry for DNA adduct formation in circumstances where exposure was accurately known, and consistent positive associations between DNA adduct formation and human cancer risk. Case–control and nested case–control epidemiological studies, examining DNA adducts in individuals exposed to a single carcinogen or carcinogenic mixtures, have shown, for many cancers, that individuals with the highest DNA adduct levels are also those with the highest cancer risk. The modest nature of these associations (odds ratio or relative risk between 1.25 and 9.10) indicates that, although DNA damage is essential, other events typically contribute to human cancer etiology.
1. Introduction
Most human exposures to chemicals that cause cancer (carcinogens — see Glossary for definitions of italicized words) occur as a result of occupation, pollution in the ambient environment, lifestyle choices, pharmaceutical use, or combinations thereof. For more than 200 years human cancer induction has been associated with exposures to chemicals or chemical mixtures. The International Agency for Research on Cancer (IARC, World Health Organization, Lyon, France) has identified approximately 100 Group 1 chemicals, defined as chemicals which, after evaluation of all the available data, are designated as carcinogenic to humans (https://www.cancer.org/cancer/cancer-causes/general-info/known-and-probablehuman-carcinogens.html). In the future, one can reasonably expect that new chemicals and/or sources of exposure will be constantly on the horizon. Furthermore, combinations of carcinogenic exposures may be more dangerous than individual single exposures. A recent example is the World Trade Center disaster of 2001, where, less than 15 years later, increased rates of thyroid and bladder cancers were reported in World Trade Center rescue and recovery workers, compared to matched residents of different states [1]. The purpose of this volume is to share knowledge that has been accumulated for many years concerning the role played by DNA damage and mutagenic events in human cancer induction. It is our anticipation that this information may assist in addressing the effects of spills, environmental disasters, and chemicals newly discovered to be carcinogenic, all of which will surely be encountered by mankind in the future.
In 1775, Sir Percival Pott, a London surgeon, published an article linking soot exposure with scrotal cancers occurring in high incidence in chimney sweeps [2]. His diagnosis that the etiology of these cancers was related to chimney soot exposure, initiated the field of occupational cancer epidemiology. He also observed that young men who spent their early years as chimney sweeps were susceptible to scrotal cancers appearing much later in life. Overall, his studies were among the first to show that human tumors arise as a result of long-term chronic carcinogen exposure, that tumors may be induced by irreversible events occurring early in life, and that reduction in exposure decreases risk of tumor formation.
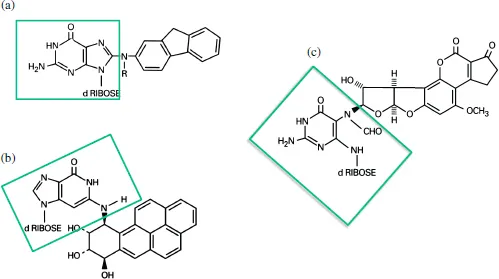
Since those early years, scientific investigations have revealed that most cancer-causing chemicals, or chemical carcinogens, act through DNA damaging or genotoxic mechanisms [3]. For example, of the hundreds of polycyclic aromatic hydrocarbons (PAHs), a major class of chemical found in soot, a small fraction are currently considered to be carcinogenic [4, 5], and one characteristic that differentiates between carcinogenic and noncarcinogenic PAHs is biotransformation to intermediates that bind to DNA. Examples of binding of a chemical to DNA, or the production of covalent addition products known as DNA adducts, are shown in Figure 1 [6]. Most cells can remove DNA adducts using DNA repair mechanisms, which both arrest DNA replication and remove the DNA damage [7]. However, unrepaired DNA adducts which remain during replication, may direct insertion of a wrong base, with subsequent formation of a mutation, or heritable change in the DNA sequence [8, 9]. Some mutations are of no consequence to the cell, but others, including those that increase proliferation (oncogenes), reduce DNA repair capacity, and/or reduce cancer protective mechanisms (tumor suppressor genes), may play critical roles in processes leading to tumorigenesis [10, 11], the essence of which is the heritable loss of growth control.
Figure 1. Molecular structures of stable carcinogen–DNA adducts formed by covalent binding with deoxyguanosine (shown in boxes) in DNA. (a) N-(deoxyguanosin-8-yl)-2-(amino)fluorene (dG-C8-AF, R=H or dG-C8- AAF, R=acetyl), formed when N-hydroxyl-aminofluorene reacts with the C8 position of deoxyguanine. (b) (7R)-N2-(10-[7β,8α,9α-trihydroxy-7,8,9,10-tetrahydro-benzo[a]pyrene]-yl)-deoxyguanosine (BPdG) is the major carcinogenic adduct formed between metabolites of benzo[a]pyrene and the exocyclic amino group of deoxyguanosine. (c) 8,9-dihydro-8-(2,6-diamino-6-formamido-4-oxo-3,4–dihydropyrimid-5-yl-amino-guanine) (AFB1-N7-G) is the major ringopened adduct of aflatoxin B1 formed with the C8 position of the deoxyguanine imidazole ring.
2. Studies in Experimental Models
In the earliest days of cancer research, it was often the discovery of human cancers [2] that led the researchers to model the same exposure and disease in animals. Tumor studies in mice and rats [12] were used to predict which chemicals might be human carcinogens, and to understand mechanisms of metabolism and other events underlying tumor incidence. These studies demonstrated dosimetry between carcinogen exposure level and tumor incidence [13], often required lifetime exposures, and revealed that cancers were specific for certain organs termed the target organs. The species examined (rat, mouse, and guinea pig), the route of exposure (oral, topical, intraperitoneal, and transplacental), and the course of exposure (weeks, months, and lifetime) were all shown to impact the tumor incidence and the target organ. However, a chemical that induced tumors in any experimental model was deemed cause for concern.
Mechanism studies showed that many chemical carcinogens, characterized as such in experimental tumor models, not only formed DNA adducts in target organs for tumorigenesis, but also formed mutations in those same target organs [14, 15]. Mechanism studies lead to the hypothesis that chemical carcinogens act by binding to DNA, inducing incorporation of a wrong base during DNA replication, and fixing that mutation so it becomes a permanent heritable trait. Early validation was found in cell culture studies, which revealed strong correlations between dose of carcinogen, formation of DNA adducts, rate of mutagenesis, and neoplastic transformation [16].
Studies in experimental models have revealed that: chronic administration of chemical carcinogens at different dose levels show dose-related increases in DNA adduct formation and tumor formation in target tissues [17]; DNA adduct formation may directly correlate with mutagenesis [18]; blocking the ability to remove/repair DNA adducts increases cancer risk [19]; and, concomitant events (inflammation, proliferation, and infection) may enhance the rate at which tumors appear [20]. Furthermore, in mouse skin studies, the mutating or initiating agent could be given at a dose insufficient to produce tumors alone, but frequent epidermal application of an inflammatory promoting agent over the lifetime of the animal would result in skin tumor growth [12].
3. DNA Damage in Humans: Methodologies
While experimental models revealed the importance of DNA damage in chemical carcinogenesis, there was little solid evidence that human cancer etiology was driven by similar mechanistic events. A mutagenic mode of action was hypothesized for most chemical carcinogens, but before about 1980 there were no methods able to measure carcinogen-induced DNA damage in human tissues. The administration and tracking of radiolabeled compounds, the classic approach used in rodents, was clearly not appropriate for use in human subjects. Without the development of new methodologies there could be no direct evidence that the paradigm for genotoxic mechanisms of chemical carcinogenesis, so elegantly elucidated in mice, existed in men.
To answer this need, multiple different methods were developed. Using an antibody-based approach for the determination of carcinogen– DNA adducts in human tissues [21], early studies revealed the presence of DNA adducts of carcinogenic PAHs in humans [22]. Over time, many different antisera were raised against either DNA modified to at least 1% by a carcinogen, or a carcinogen–DNA adduct covalently bound to a carrier protein. These antisera typically cross-reacted with a single DNA adduct, or a famil...