
eBook - ePub
Clinical Handbook of Interstitial Lung Disease
Keith C. Meyer, Muhunthan Thillai, David R. Moller, Muhunthan Thillai, David R. Moller, Keith C. Meyer
This is a test
Partager le livre
- 529 pages
- English
- ePUB (adapté aux mobiles)
- Disponible sur iOS et Android
eBook - ePub
Clinical Handbook of Interstitial Lung Disease
Keith C. Meyer, Muhunthan Thillai, David R. Moller, Muhunthan Thillai, David R. Moller, Keith C. Meyer
Détails du livre
Aperçu du livre
Table des matières
Citations
À propos de ce livre
This handbook provides clinical guidance to the practicing physician on the diagnosis and treatment of Interstitial Lung Diseases (ILD). A contributed work with invited chapters which draw on the knowledge and experience of recognised global leaders in respiratory medicine, it is authoritative, concise and portable and is intended for use in a fast-paced clinical setting. The book:
- offers practical tips and clear guidance for clinicians
-
- provides detailed explanations of the main therapeutic options for each individual ILD
-
- contains high-quality visuals, including radiology and histopathology of the most common as well as some of the rarer ILDs
-
- discusses individual ILDs and has topics common to all including critical care, lung transplantation and palliative care
-
- navigates clinicians through cases with decision making guidelines and algorithms
-
- includes appendices with international practice guidelines, sample patient information sheets and other helpful resources.
-
Emphasizing how to perform a thorough assessment of an ILD patient for accurate diagnosis and their subsequent effective management, this is both a gold standard text as well as a daily companion for physicians caring for ILD patients. A first-of-its-kind, it will become the go-to guide for all clinicians who manage patients with ILD.
Foire aux questions
Comment puis-je résilier mon abonnement ?
Il vous suffit de vous rendre dans la section compte dans paramètres et de cliquer sur « Résilier l’abonnement ». C’est aussi simple que cela ! Une fois que vous aurez résilié votre abonnement, il restera actif pour le reste de la période pour laquelle vous avez payé. Découvrez-en plus ici.
Puis-je / comment puis-je télécharger des livres ?
Pour le moment, tous nos livres en format ePub adaptés aux mobiles peuvent être téléchargés via l’application. La plupart de nos PDF sont également disponibles en téléchargement et les autres seront téléchargeables très prochainement. Découvrez-en plus ici.
Quelle est la différence entre les formules tarifaires ?
Les deux abonnements vous donnent un accès complet à la bibliothèque et à toutes les fonctionnalités de Perlego. Les seules différences sont les tarifs ainsi que la période d’abonnement : avec l’abonnement annuel, vous économiserez environ 30 % par rapport à 12 mois d’abonnement mensuel.
Qu’est-ce que Perlego ?
Nous sommes un service d’abonnement à des ouvrages universitaires en ligne, où vous pouvez accéder à toute une bibliothèque pour un prix inférieur à celui d’un seul livre par mois. Avec plus d’un million de livres sur plus de 1 000 sujets, nous avons ce qu’il vous faut ! Découvrez-en plus ici.
Prenez-vous en charge la synthèse vocale ?
Recherchez le symbole Écouter sur votre prochain livre pour voir si vous pouvez l’écouter. L’outil Écouter lit le texte à haute voix pour vous, en surlignant le passage qui est en cours de lecture. Vous pouvez le mettre sur pause, l’accélérer ou le ralentir. Découvrez-en plus ici.
Est-ce que Clinical Handbook of Interstitial Lung Disease est un PDF/ePUB en ligne ?
Oui, vous pouvez accéder à Clinical Handbook of Interstitial Lung Disease par Keith C. Meyer, Muhunthan Thillai, David R. Moller, Muhunthan Thillai, David R. Moller, Keith C. Meyer en format PDF et/ou ePUB ainsi qu’à d’autres livres populaires dans Médecine et Médecine pulmonaire et thoracique. Nous disposons de plus d’un million d’ouvrages à découvrir dans notre catalogue.
Informations
1
An overview of the classification and diagnosis of interstitial lung disease
Keith C Meyer
Historical background and evolution of terminology
Current classification systems for ILD
Idiopathic interstitial pneumonias
Genetics of ILD: Potential impact on classification and terminology
Current approach to making a confident ILD diagnosis
Summary
References
Historical background and evolution of terminology
Recognition of the existence of interstitial lung disease (ILD) dates back more than 100 years when Sir William Osler described ‘cirrhosis of the lungs’ and recognized the diversity of its forms and the difficulty of classifying these disorders (1). Various terminologies were coined over the span of the twentieth century as various leaders in the field described patients whose lungs displayed changes of interstitial inflammation and fibrosis. Hamman and Rich (1944) described four cases of rapidly progressive, diffuse alveolar wall thickening without identifiable cause, which led to use of the term, ‘Hamman–Rich syndrome’ for either acute-onset or chronic fibrotic ILD. Subsequently, diffuse pulmonary fibrosis was linked to forms of connective tissue disease (CTD) and other causes, such as exposure to organic or inorganic dusts and pneumotoxic drug reactions, but many forms remained unexplained by any associations. Terms such as ‘chronic idiopathic interstitial fibrosis’, ‘diffuse fibrosing alveolitis’ or ‘idiopathic pulmonary fibrosis’ were used to designate fibrotic ILD of unknown aetiology, and these disorders were thought to occur as a consequence of alveolar wall inflammation (‘alveolitis’). The term ‘diffuse fibrosing alveolitis’ was coined by Scadding (2) to describe widespread fibrotic change beyond the level of the terminal bronchioles, and Scadding subdivided entities according to known or unknown associations and patterns of fibrosis (Figure 1.1). Liebow and Carrington (3,4) published a classification system for chronic idiopathic interstitial pneumonias that was based on histopathologic changes; one of the five subgroups that they described was termed ‘usual’ interstitial pneumonia (UIP), and the Hamman–Rich syndrome was felt to be an acute form of UIP (Figure 1.1).
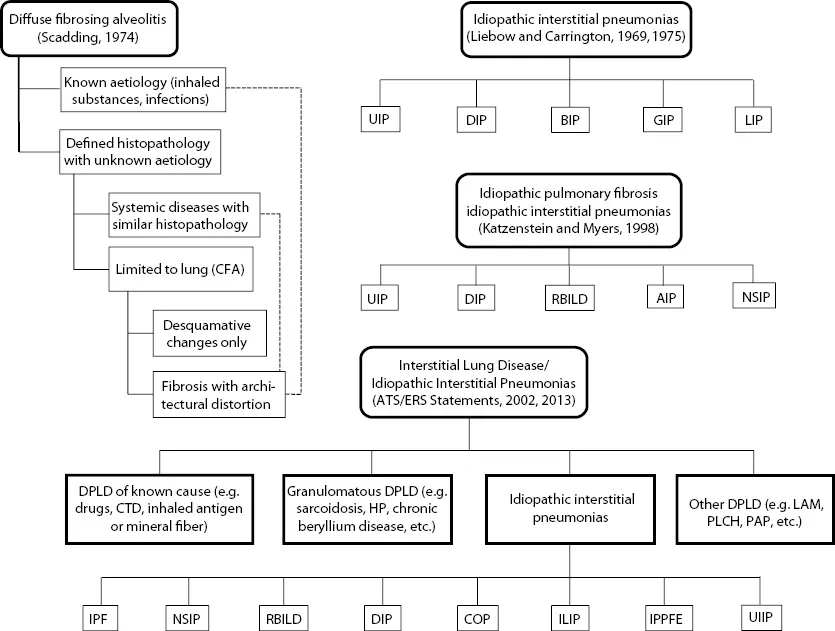
Figure 1.1Evolution of classification systems for interstitial lung disease and forms of idiopathic interstitial pneumonia. (Abbreviations: AIP = acute interstitial pneumonia; BIP = bronchiolitis interstitial pneumonia; CFA = cryptogenic fibrosing alveolitis; COP = cryptogenic organizing pneumonia; CTD = connective tissue disease; DIP = desquamative interstitial pneumonia; DPLD = diffuse parenchymal lung disease; GIP = giant cell interstitial pneumonia; HP = hypersensitivity pneumonia; ILIP = idiopathic lymphoid interstitial pneumonia; IPF = idiopathic pulmonary fibrosis; IPPFE = idiopathic pleuropulmonary fibrosis; LAM = lymphangioleiomyomatosis; NSIP = non-specific interstitial pneumonia; PLCH = pulmonary Langerhans cell histiocytosis; RBILD = respiratory bronchiolitis interstitial lung disease; UIIP = undifferentiated interstitial pneumonia; UIP = usual interstitial pneumonia.)
The classification systems proposed by Scadding or Liebow and Carrington were quite similar, but clinicians tended to overlook histopathologic variations and termed ‘chronic idiopathic fibrosing ILD’ as ‘cryptogenic fibrosing alveolitis’ (Europeans) or ‘idiopathic pulmonary fibrosis’ (United States). Katzenstein and Myers (5) re-examined Liebow’s classification system and refined the histopathology-based system by adding two new categories (non-specific interstitial pneumonia [NSIP] and respiratory-bronchiolitis-associated ILD [RBILD]) while revising/retaining some of Liebow’s categories (UIP and desquamative interstitial pneumonia [DIP]): acute interstitial pneumonia (AIP) was coined as a term for the Hamman–Rich syndrome (Figure 1.1). Their scheme recognized giant cell interstitial pneumonia (GIP) as caused by hard-metal exposure and considered lymphoid interstitial pneumonia (LIP) as a lymphoproliferative disorder, while bronchiolitis interstitial pneumonia (BIP) was recognized as an intraluminal (rather than interstitial) process that could take the form of organizing pneumonia (aka bronchiolitis obliterans organizing pneumonia [BOOP]) or diffuse alveolar damage (DAD). They also correlated their histopathologic pattern-based classification system with clinical features and natural history.
With the evolution of ongoing clinical and pathologic investigations of ILD from the late twenti...