![]()
John Wiley & Sons, Inc.
Wiley Interdisciplinary Reviews: Systems Biology and Medicine
© 2010 John Wiley & Sons, Inc
DOI: 10.1002/wsbm.124
Advanced Review
Mediators and dynamics of DNA methylation
Robert Shoemaker,1 Wei Wang1 and Kun Zhang2*
1Department of Chemistry and Biochemistry, University of California at San Diego, La Jolla, CA, USA
2Department of Bioengineering, University of California at San Diego, La Jolla, CA, USA
As an inherited epigenetic marker occurring mainly on cytosines at CpG dinucleotides, DNA methylation occurs across many higher eukaryotic organisms. Looking at methylation patterns genome-wide classifies cell types uniquely and in several cases discriminates between healthy and cancerous cell types. DNA methylation can occur allele-specifically, which allows the cellular regulatory machinery to recognize each allele separately. Although only a small number of allele specifically methylated (ASM) regions are known, genome-wide experiments show that ASM is prevalent throughout the human genome. These DNA methylation patterns can be modified via DNA demethylation, which is important for induced pluripotent stem reprogramming and primordial germ cells. Recent evidence shows that the protein activation-induced cytidine deaminase plays a critical role in these demethylation events. Many transcription factors mediate DNA methylation patterns. Some transcription factors bind specifically to methylated or unmethylated sequences and other transcription factors protect genomic regions (e.g., promoter regions) from nearby DNA methylation encroachment. Possibly acting as another epigenetic regulatory layer, methylated cytosines are also converted to 5-hydroxyethylcyotines, which is a new modification type whose biological significance has yet been defined. © 2010 John Wiley & Sons, Inc. WIREs Syst Biol Med 2011 3 281–298 DOI: 10.1002/wsbm.124
For further resources related to this article, please visit the WIREs website
INTRODUCTION
DNA methylation is an inherited epigenetic chemical modification that occurs primarily on the cytosines in CpG dinucleotides. However, examples of non-CpG methylation are found in plants and human embryonic stem cells.1–3 The proteins DNA Methyltransferase 1 (DNMT1), DNMT3a, and DNMT3b are known to methylate cytosines, while protein families, including Methyl-CpG Binding Domain (MBD) containing proteins, SET and RING finger-associated (SRA) domain containing proteins, and other zinc finger proteins, recognize the presence of methylated cytosines.4–13 DNMT1 maintains methylation patterns and this maintenance is required for normal cell division.14–16 Originally thought to bind DNA due to its similar structure and sequence to other members within the DNMT family, DNMT2 was later found to specifically target RNA.14,17,18 DNMT3a and DNMT3b have de novo methylation activity that is required for embryonic development.19 Another protein, DNMT3L, colocalizes with DNMT3a and DNMT3b, and it enhances de novo methylation in vitro and in vivo.20–26 DNMT3L itself lacks methyltransferase activity and thus enhances methylation via its interaction with DNMT3a. DNA methylation is widespread across many organisms, including bacteria, insects, and mammals, and it is most commonly associated with transcriptional silencing.27–30 As many repetitive regions and transposons are methylated in various genomes, DNA methylation is thought to act as a genomic defense mechanism that prevents the activation of these sequences.31–34 DNA methylation is also present in genes and regulatory regions, where it is correlated with transcriptional repression or activation depending on its context. Given the regulatory effects associated with DNA methylation, characterizing the methylation states of cytosines genome-wide reveals the biological state of the host cell on a global scale.1–3,35–37 This article (1) discusses the biological meaning of DNA methylation patterns on intercellular and intracellular levels, (2) explores the importance of the mechanisms that erase these patterns, (3) discusses the role of transcription factors in DNA methylation, and (4) reviews the presence of a new cytosine modification, hydroxymethylcytosine, which is a product of DNA methylation.
INTERCELLULAR DNA METHYLATION SIGNATURES: CONSERVATION AND APPLICATIONS
DNA Methylation in Higher Eukaryotes
A comparison of DNA methylation patterns across eight species (Arabidopsis, green algae, rice, poplar, honey bee, mouse, sea squirt, and zebrafish) revealed not only many common features but also striking differences.27 Repetitive regions and transposons were enriched for CpG methylation relative to nearby regions for all eight organisms, but these methylation patterns in sea squirt were much weaker. Regions of high CpG density, which are known as CpG islands, are mostly unmethylated in vertebrates (i.e., zebrafish and mouse) but overall vertebrate methylation levels are high (70–80% global CpG methylation). Non-CpG (CHG and CHH) methylation was found in all three flowering plants; CHG and CHH methylation was enriched at transposons and repetitive regions. CHH methylation describes a methylated cytosine followed by two nucleotides that may not be guanine. CHG methylation entails a methylated cytosine that precedes an adenine, thymine, or cytosine, followed by guanine. Unlike CG or CHG methylation, CHH sites are strand specific, which means, for example, a CHH site on the Watson strand is not found on the reverse complementary Crick strand. Although exhibiting non-CpG methylation, green algae’s CHH and CHG patterns showed no enrichment in any particular genomic region. The remaining organisms displayed a much lower percentage of non-CpG methylation. Vertebrates did not show a significant enrichment of CpG methylation in exons relative to introns as was seen in all three flowering plants. The honeybee, although displaying low levels of global CpG methylation, displayed significant CpG methylation in exon regions. Green algae showed very low global CpG methylation, but exons were more methylated. Sea squirt also showed preferential methylation of exon regions. This conservation study shows that DNA methylation is present in higher eukaryotes and that DNA methylation often targets certain genomic regions. However, the targeted regions are not entirely consistent across all of the eight studied organisms.
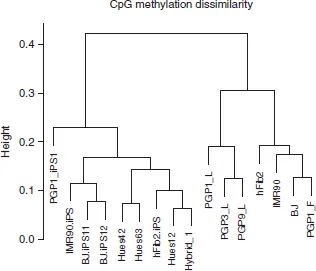
Since CpG methylation sites are known in a sequenced genome, one can easily compare methylation signatures across intraspecies cell lines. Genome-wide DNA methylation patterns are cell type specific. Our group targeted bisulfite sequencing of CpG islands across chr12 and chr20, and found that the methylation frequency data clearly distinguished between human embryonic stem cell lines, fibroblasts, and lymphoblasts38,39 (Figure 1). An additional study reported a conserved set of regions that are uniquely methylated between human liver, spleen, and brain tissues.40 These differentially methylated regions tended to be located just outside of CpG islands, and were thus called CpG island shores. Methylation of these CpG island shores had a strong inverse relationship with the expression of associated genes. The authors labeled 16,379 regions as differentially methylated across examined tissues (T-DMRs), and the median length of these regions was 255 bp. Extending this analysis to 13 colorectal cancer cell samples with matched normal mucosal samples, the authors found a separate set of differentially methylated regions (C-DMRs) that showed significant differences in methylation between the normal mucosal and the matched cancer samples. Forty-five percent of the 2707 identified C-DMRs overlapped with T-DMRs (P-value < 10−14), which showed that these many of these DMRs could discriminate between tissues and colorectal cancer. A continuation of this study found 4401 regions were differentially methylated in iPS cells relative to the untransformed fibroblast cells (R-DMRs).41 These R-DMRs, similar to the C-DMRs and T-DMRs, overlapped significantly with CpG island shore regions (over 70%). Many R-DMRs also overlapped with T-DMRs (56%). These studies showed that DNA methylation signatures are unique across many different types of cell lines.
DNA Methylation as a Clinical Biomarker
DNA methylation can classify cell types into subpopulations, which can exhibit unique phenotypes. One cancer study examined the methylation profiles of blast cells taken from 344 diagnosed acute myeloid leukemia (AML) patients.42 Clustering these methylation profiles created 16 unique AML subtype clusters. Three of those patient clusters were defined by the WHO classification,43 eight were enriched for specific genetic or epigenetic lesions, and the remaining five could not be explained by current knowledge; all of these subtypes were distinct when compared to normal bone marrow cells. The authors used the methylation signatures of 18 methylation probe sets that covere...