![]()
Part 1
General Aspects
![]()
1
An Overview of the Discovery and Development Process for Biologics
Heather H. Shih, Paula Miller and Douglas C. Harnish
1.1 Introduction
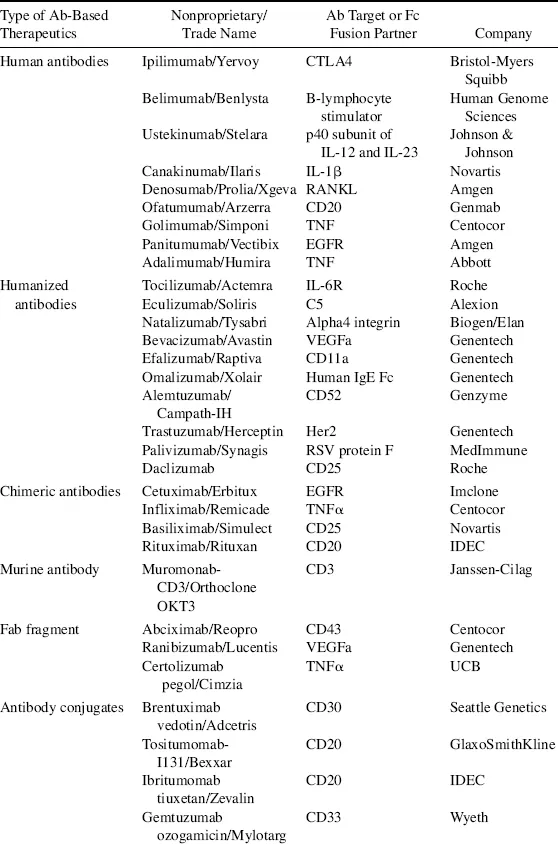
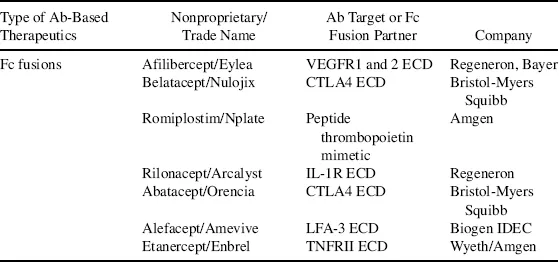
Biologics, also called biotherapeutics or biopharmaceuticals, are drug substances derived from living organisms or produced using biotechnology that are composed of biological entities such as proteins, peptides, nucleic acids, or cells [1]. They differ from small molecule (SM) drugs that are chemically synthesized and have low molecular weights. Some biologics, such as antibody–drug conjugates, consist of both a protein moiety and an SM component, both of which are required for the therapeutic action of the drug. Traditional biologics that have reached the market include vaccines and blood-derived factors. The advancement in modern biotechnology has brought forth new classes of biologics as exemplified by monoclonal antibodies (mAbs), Fc fusion proteins, recombinant proteins, and peptide drugs. Some early clinical success is now seen in several novel classes of biologics, which include antibody variants, novel protein scaffolds, RNA therapeutics, and cell-based therapies [2–5]. This chapter focuses on protein-based biologics, particularly mAbs because they represent the largest class of biologic drugs. By the end of 2011, the US Food and Drug Administration (FDA) had approved close to 40 mAbs and antibody variants as summarized in Table 1.1. Details on other forms of biologics such as vaccines and RNA drugs can be found in later chapters.
Table 1.1 List of Food and Drug Administration–Approved Antibody-Based Therapeutics Up to 2011 as Categorized by Types.
The first protein-based biologic drug, recombinant insulin Humulin, was approved in the United States in 1982 [6]. Since then the field of biologics grew steadily, with the biotechnology sector laying the foundation for both the drug discovery process and technology innovation. Around late 1990s, the pharmaceutical industry started to invest more in the development of biologics. This shift from a primary focus on SM drugs was largely due to patent expiration on these drugs and the concurrent fierce competition from generic SM drugs. In addition, the increasing difficulty to bring new drugs to the market because of tightened regulations and a lack of breakthroughs in the drug discovery process has also contributed to this shift. Presently, the number of biologics on the market has reached more than 200, and the sales of biologics in 2009 reached $93 billion, with approximately one third of current pharmaceutical pipelines consisting of biologics [7]. Given that almost all of the large pharmaceutical companies have acquired infrastructures and committed resources to develop biologics, we will continue to see a robust growth in this sector in the coming years.
Compared with SM drugs, protein-based biologics have unique therapeutic features. A therapeutic protein usually exhibits exquisite specificity when binding to and modulating its molecular target, which often translates into low off-target toxicity and clinical safety. For example, therapeutic mAbs bind to their target molecules with affinities in the picomolar to low nanomolar range (e.g., [8]). Furthermore, the interaction occurs over a broad interface with multiple physical and chemical bonds formed between an antibody and its cognate antigen, resulting in an extraordinary binding specificity that allows the differentiation of binding partners that differ by as few as one amino acid or subtle conformational difference. On the contrary, the small size of an SM drug makes it prone to off-target binding to proteins other than its intended target, which may result in unacceptable levels of toxicities. A potentially short development cycle is another advantage for the development of biologics, particularly mAbs and recombinant proteins. A clinical candidate for mAb or recombinant protein can be generated and selected in as short as 3 to 5 years compared with typically 7 to 8 years for SMs.
Protein-based biologics have their own limitations. Presently, almost all protein-based drugs must be administered as intravenous or subcutaneous injections because oral delivery is not yet a viable route of administration. Furthermore, protein drugs do not readily penetrate cell membrane and blood–brain barrier (BBB) and therefore are limited to the modulation of peripherally located extracellular targets. The cost of goods to manufacture protein drugs is significantly higher than for SM drugs, which translates into a high drug price that exacerbates health management cost issues [9].
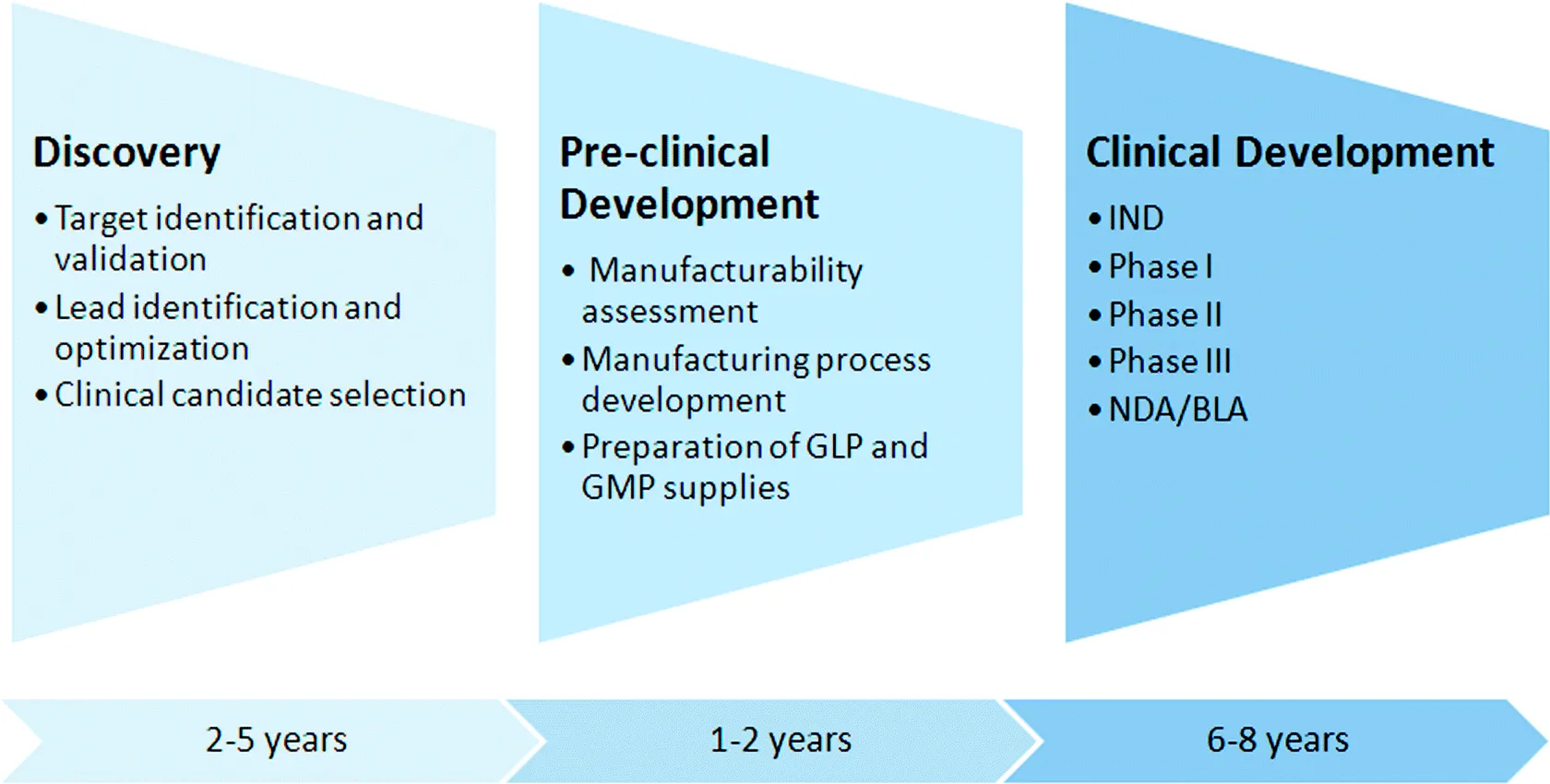
Based on these pros and cons associated with the development of biologics, presently the pharmaceutical industry strives to achieve a balanced portfolio consisting of both SM and biologic drugs. This chapter provides an overview of the discovery and development process for protein therapeutics with a primary focus on mAbs (Fig. 1.1). Additionally, the chapter summarizes the current status of the protein-based biologics field and discusses several future trends.
1.2 The Discovery Process for Monoclonal Antibodies
Monoclonal antibodies and mAb variants such as antibody–drug conjugates and Fc fusion proteins are a major class of biologics. This section describes in detail the discovery process for mAbs. Later chapters illustrate the process for several other forms of biologics such as vaccines and RNA therapeutics.
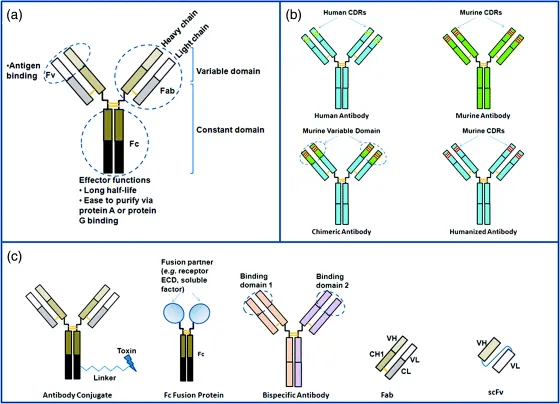
In mammals, antibodies are proteins found in the blood that are produced by B cells from the humoral immune system in defense of foreign organisms recognized by the host system. Also known as immunoglobulins, antibodies can be categorized into five classes or isotypes, namely IgM, IgD, IgG, IgE, and IgA (reviewed by Schroeder and Cavacini [10]). IgG is the predominant class in therapeutic antibodies. An IgG molecule consists of two heavy chains and two light chain interlinked by disulfide bonds (Fig. 1.2). Each chain has an N-terminal variable region (Fv) and a C-terminal constant region. The variable region of each pair of heavy and light chains has six hypervariable loops (three per chain) called complementarity determining regions (CDRs) that form the antigen binding region at the tip of the IgG molecule. The heavy and light chains in an Fv fragment can be joined via a linker using recombinant DNA technology and produced as a single-chain Fv (scFv) molecule, which is used in phage display technology as a method to generate therapeutic antibody candidates (see later discussion). Papain digestion of an IgG yields three components: two Fab fragments and an Fc fragment. A Fab molecule can be generated via molecular engineering into a therapeutic agent. Currently, three Fab-based antibody drugs are on the market: abciximab, ranibizumab, and certolizumab pegol (Table 1.1). The Fc fragment exhibits effector functions, namely the ability to engage immune system to kill antibody opsonized molecules. In addition, the binding of Fc to bacterial protein A and protein G has been applied to the purification of therapeutic antibodies at industrial manufacturing scales. Furthermore, Fc binds to the neonatal Fc receptor (FcRn) expressed on endothelial cells. Upon uptake by endothelial cells, FcRn recycles IgG molecules back into circulation, thus conferring a long in vivo half-time to IgG [11]. Via molecular engineering, the Fc fragment can be fused to another protein fragment such as a soluble factor or the extracellular domain of a cell surface receptor. The resulting Fc fusion proteins represent one type of antibody-based therapeutics (Table 1.1 and Fig. 1.2).
The advancement in molecular and cellular biology has transformed the isolation, molecular engineering, and production of recombinant mAbs into an industrial drug-making process. Compared with other protein drugs, mAbs demonstrate superior properties as therapeutic molecules. They typically exhibit exquisite specificity to their molecular targets and minimal off-targeting binding. The bivalency of each antibody molecule contributes to its extraordinarily high binding strength (avidity) as the summation of the affinity from each half of the molecule. It also confers a cross-linking function that can be applied to its therapeutic function. The effector functions associated with the Fc domain allow the molecule to effectively mobilize the immune system to attack and kill tumor cells when used to treat cancers. mAbs are highly stable proteins with natural resistance to biological and chemical degradation. They tend to be amenable to expression and purification at manufacturing scales. They typically exhibit long in vivo half-lives, which allow infrequent administration in patients. As a result of these multifaceted advantages associated with mAbs, the pharmaceutical industry has focused on the development of mAbs as a major class of biologic drugs.
In many pharmaceutical and biotechnology companies, the development process for therapeutic mAbs is well established and analogous to that for SM drugs. It can be generalized into four stages: target selection, screening preparation, lead selection and optimization, and clinical candidate selection. The discovery phase is followed by a preclinical development process and ultimately the clinical testing of the selected mAb candidate in human subjects. This section describes relevant research activities that take place in each of the four discovery stages leading to the selection of a clinical candidate protein. The major differences in the respective processes for developing biologics and SMs are discussed at the end of this section.
1.2.1 Target Selection
A target is a biological entity in patients that can be specifically and effectively modulated by a drug to ameliorate or cure a pathological condition. Selecting a drug target is usually the first step in a drug development program, although there are exceptions to the rule when drug candidate screening is carried out using a functional readout without predefined targets. In a majority of such cases, the target is identified retrospectively after the functional candidates have been selected.
A novel target is often identified either during the studies of biological pathways underlying a disease or as a result of disease target identification efforts frequently using genomic technologies such as transcriptional profiling, proteomics, and genome-wide gene association studies. Usually a target must be “validated” before the initiation of a drug discovery program. Target validation refers to a process of collecting clinical and experimental data to predict a beneficial therapeutic outcome from a hypothesized modulation ...