![]()
CHAPTER 1
SEPARATION MECHANISMS IN HYDROPHILIC INTERACTION CHROMATOGRAPHY
DAVID V. MCCALLEY
Department of Applied Sciences, University of the West of England, Bristol, UK
1.1 Introduction
1.2 Historical Background: Recognition of the Contribution of Partition, Ion Exchange, and RP Interactions to the Retention Process
1.3 Recent Studies on the Contributory Mechanisms to HILIC Retention 1.3.1 Overview
1.3.2 Contribution of Adsorption and Partition to HILIC Separations
1.3.3 Further Studies on the Contribution of Ionic Retention in HILIC
1.3.4 RP Retention on Bare Silica
1.3.5 Electrostatic Repulsion Hydrophilic Interaction Chromatography (ERLIC): A New Separation Mode in HILIC
1.4 Conclusions
1.1 INTRODUCTION
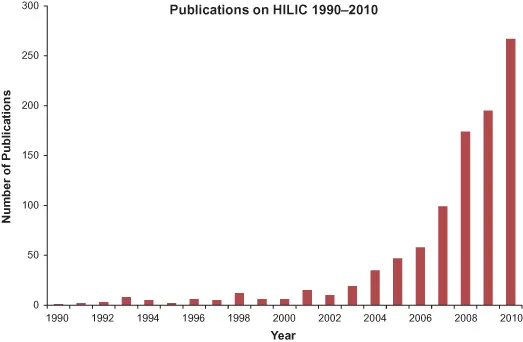
Hydrophilic interaction chromatography (HILIC) is a technique that has become increasingly popular for the separation of polar, hydrophilic, and ionizable compounds, which are difficult to separate by reversed-phase chromatography (RP) due to their poor retention when RP is used. HILIC typically uses a polar stationary phase such as bare silica or a polar bonded phase, together with an eluent that contains at least 2.5% water and >60% of an organic solvent such as acetonitrile (ACN). However, these values should not be regarded as definitive of the rather nebulous group of mobile and stationary phase conditions that are considered to constitute HILIC. Figure 1.1 shows the number of publications on HILIC between the years 1990 (when the term was first employed) and 2010 according to the Web of Knowledge [1] using the search terms “HILIC” or “hydrophilic interaction (liquid) chromatography.” For the first 12 years or so, the number of publications remained between 1 and 15, but after this period, interest increased rapidly from 19 publications in 2003 to 267 in 2010. While HILIC has unique retention characteristics for hydrophilic compounds, this increase in interest also reflects the advantages of HILIC over RP methods in situations where either technique is applicable. These advantages result mostly from the high organic content of typical mobile phases and their resultant high volatility and low viscosity. A particular advantage is in coupling HILIC to mass spectrometry (MS) as mobile phases are more efficiently desolvated in interfaces such as electrospray, giving rise to better sensitivity than with RP methods. Thus, Grumbach and coworkers demonstrated sensitivity increases of 3–4 orders of magnitude when comparing the analysis of the drugs salbutamol and bamethan by HILIC on a bare silica column using a gradient analysis starting at 90% ACN with that on a C18 RP column using a gradient starting at 0% ACN [2]. Columns can be used at considerably lower pressures than in RP; the viscosity of 80–90% ACN mixtures with water as typically used in HILIC is only about half that of 20–30% ACN mixtures that might be used in RP separations [3]. Alternatively, longer columns can be used at pressures typically found in RP analysis, allowing high efficiencies to be obtained [4]. For example, when combining the low viscosity of HILIC with the efficiency gains shown by superficially porous (shell) particle columns, it is possible to generate column efficiencies in excess of 100,000 plates with reasonable analysis times, and using pressures that are well within the capabilities of conventional HPLC systems (pressure < < 400 bar). Low viscosity also results in increased solute diffusion in the mobile phase, giving rise to smaller van Deemter C terms and improved mass transfer, and the possibility of operating columns at high flow rates with reduced losses in efficiency for fast analysis [5]. Surprisingly good peak shapes can be obtained for some basic compounds. For example, efficiencies of around 100,000 plates/m with asymmetry factors (As) close to 1.0 were reported for basic drugs such as nortritpyline (pKa ∼10) using a 5-µm particle size bare silica HILIC phase. In comparison, such solutes often give rise to peak asymmetry in RP separations.
A separate advantage of HILIC is its compatibility with sample preparation methods using solid-phase extraction (SPE). Some such methods incorporate an elution step that uses a high concentration of an organic solvent, which gives rise to a potential injection solvent of the eluate that is stronger than typical RP mobile phases [2]. This mismatch in solvent strengths can give rise to peak broadening or splitting, necessitating evaporation of the SPE eluate and reconstitution in the mobile phase. SPE eluates with high organic solvent concentrations can be injected directly in HILIC, as they are weak solvents in this technique. The combination of different retention mechanisms in sample purification and analysis steps (HILIC/RP) can be advantageous in giving extra selectivity compared with an RP/RP procedure, where in some cases the SPE column may act merely as a sort of filter for the analytical column [6].
While HILIC is simple to implement in practice, some recent papers have concluded that the separation mechanism is a complex multiparametric process that may involve partition of solutes between a water layer held on the surface and the bulk mobile phase, adsorption via interactions such as hydrogen bonding and dipole–dipole forces, ionic interactions, and even nonpolar retention mechanisms (similar to RP interactions), depending on the stationary and mobile phases [7–9]. In this chapter, we will consider in some detail the various mechanisms that contribute to HILIC separations.
1.2 HISTORICAL BACKGROUND: RECOGNITION OF THE CONTRIBUTION OF PARTITION, ION EXCHANGE, AND RP INTERACTIONS TO THE RETENTION PROCESS
The term “hydrophilic interaction chromatography” was coined in 1990 by Alpert [10]. He carefully avoided the acronym HIC to avoid confusion with the technique of hydrophobic interaction chromatography, the latter being an adaptation of the RP technique where decreasing salt concentrations are used to progressively elute large biomolecules from the stationary phase. However, it is possible that HILIC dates back to the earliest days of liquid chromatography, when Martin and Synge separated amino acids on a silica column using water-saturated chloroform as the mobile phase. These authors explained the separation mechanism as being the partitioning of the solutes between a water layer held on the column surface and the chloroform [11]. The silica was considered to act merely as a mechanical support. It later became clear that use of a solvent that is immiscible with water, such as chloroform, is not an essential requirement. Lindon and Lawhead [12] discussed the separation of sugars such as fructose, glucose, sucrose, melibiose, and raffinose on a micro-Bondapak carbohydrate column (an aminosilica column, 10 µm particle size). The mobile phase was ACN–water (75:25, v/v); the authors showed that increasing the concentration of water reduced the retention times of the sugars. The authors noted that while the α- and β-anomers of sugars are readily separated by gas-liquid chromatography, they were not separated by this LC method, removing an unnecessary complication. However, no explanation for this lack of separation, or for the retention mechanism, was presented. It was shown later that aminopropyl silica in the presence of ACN–water greatly increased the mutarotation rate of the sugars compared with the effect of bare silica [13]. This effect is due to the basic environment generated in the column pores by the presence of the amino groups [14]. With a refractive index detector, it was shown that water was retained on the aminopropyl silica when pumping mobile phases of ACN–water and that the volume fraction of water in the liquid associated with the stationary phase was much higher than that in the corresponding eluent. The extent of water enrichment in the stationary liquid was found to be relatively high when the eluent contained a low water concentration. The separation of the sugars was explained as being due to their partition between the water-rich liquid in the stationary phase and the bulk mobile phase. Using a similar experimental procedure, other workers showed a reduced uptake of water on an aminosilica column when methanol–water was used as the mobile phase compared with ACN–water, as the competition between water and methanol for polar sites on the column was increased [15].
While the reports on sugar analysis were clearly classical HILIC separations in their use of a polar phase together with an ACN–water mobile phase containing a high concentration of organic solvent, a number of other early papers using bare silica columns demonstrated separations that contain at least some of the mechanisms that are now considered contributory to HILIC. Bidlingmeyer and coworkers [16] separated organic amines on a silica column using “reversed-phase eluents” consisting typically of ACN–water (60:40, v/v) containing ammonium phosphate buffer, pH 7.8. They showed that increasing the salt concentration decreased the retention of ionized basic compounds, indicating the contribution of ionic retention to the overall mechanism. It was demonstrated that over the range 70–30% ACN, if buffer strength and pH were held constant, retention decreased with increasing proportion of ACN as would be expected in a reversed-phase separation. Good peak symmetry was obtained for basic compounds on these bare silica columns. The authors concluded from a comparison with RP that the key to good peak shapes with these solutes was not the presence or absence of silanols but more probably the accessibility of these surface groups. Nevertheless, the concentrations of ACN employed in this work were at the lowest end of the range generally used for HILIC separations, and it is questionable whether the important partition element of the HILIC mechanism was involved to any extent in such separations, as the mobile phase becomes more hydrophilic and thus competitive with the stationary phase. Other early work by Flanagan and Jane also showed the separation of basic drugs on bare silica columns, but this time using nonaqueous ionic eluents [17,18]. The nonaqueous, primarily methanolic eluents, contained additives such as perchloric acid or ammonium perchlorate of appropriate pH and ionic strength. The authors demonstrated that the retention of quaternary compounds increased with eluent pH, particularly in the pH range of 7–9, whereas the retention of base...