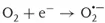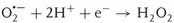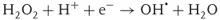Water is one of the essential resources on our planet. Therefore, fresh water and the recycling of waste-water are very important topics in various areas. Energy-saving green technologies are a demand in this area of research.
Photocatalysis comprises a class of reactions which use a catalyst activated by light. These reactions include the decomposition of organic compounds into environmental friendly water and carbon dioxide, leading to interesting properties of surfaces covered with a photocatalyst: they protect e.g. against incrustation of fouling matter, they are self-cleaning, antibacterial and viricidal. Therefore, they are attractive candidates for environmental applications such as water purification and waste-water treatment.
This book introduces scientists and engineers to the fundamentals of photocatalysis and enlightens the potentials of photocatalysis to increase water quality. Also, strategies to improve the photocatalytic efficacy are pointed out: synthesis of better photocatalysts, combination of photocatalysis with other technologies, and the proper design of photocatalytic reactors. Implementation of applications and a chapter on design approaches for photocatalytic reactors round off the book.
'Photocatalysis and Water Purification' is part of the series on Materials for Sustainable Energy and Development edited by Prof. G.Q. Max Lu. The series covers advances in materials science and innovation for renewable energy, clean use of fossil energy, and greenhouse gas mitigation and associated environmental technologies.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Fundamentals: Active Species, Mechanisms, Reaction Pathways
1
Identification and Roles of the Active Species Generated on Various Photocatalysts
Yoshio Nosaka and Atsuko Y. Nosaka
TiO2 photocatalysts have been utilized for the oxidation of organic pollutants [1–5]. For further practical applications, the improvement in the photocatalytic efficiency and the extension of the effective wavelength of the irradiation light are desired. From this point of view, better understanding of the primary steps in photocatalytic reactions is prerequisite to develop prominent photocatalysts. The properties of TiO2 and the reaction mechanisms in molecular level have been reviewed recently [6]. Therefore, this chapter describes briefly active species involved in the photocatalytic reactions for bare TiO2 and TiO2 modified for visible-light response, that is, trapped electrons, superoxide radical (O2⋅−), hydroxyl radical (OH⋅), hydrogen peroxide (H2O2), and singlet oxygen (1O2).
1.1 Key Species in Photocatalytic Reactions
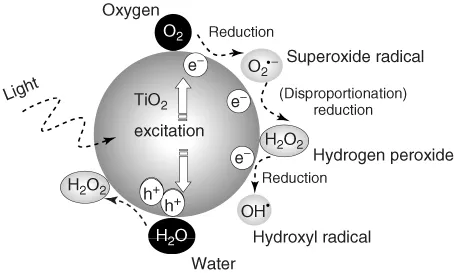
Since the photocatalytic reactions proceed usually with oxygen molecules (O2) in air, the reduction of oxygen would be the important process in photocatalytic reduction. On the other hand, taking into account that the surface of TiO2 photocatalysts is covered with adsorbed water molecules in usual environments and that photocatalysts are often used to decompose pollutants in water, oxidation of water would be the important process in photocatalytic oxidation. As shown in Figure 1.1, when O2 is reduced by one electron (Eq. (1.1)), it becomes a superoxide radical (O2⋅−) that is further reduced by one electron (Eq. (1.2)) or reacts with a hydroperoxyl radical (HO2⋅, i.e., protonated O2⋅−) to form hydrogen peroxide (H2O2). The latter reaction is largely pH dependent because the amount of HO2⋅, whose pKa is 4.8, changes largely at pH around neutral [7]. One-electron reduction of H2O2 (Eq. (1.3)) produces hydroxyl radical (OH⋅). In the field of radiation chemistry, it is well documented that OH⋅ is produced by one-electron oxidation of H2O with ionization radiation. However, the formation of OH⋅ in the photocatalytic oxidation process has not been confirmed, as described later.
1.1
1.2
1.3
Figure 1.1 One-electron reduction steps of oxygen to OH radical and two-electron oxidation step of water to H2O2 observed in the TiO2 photocatalyst.
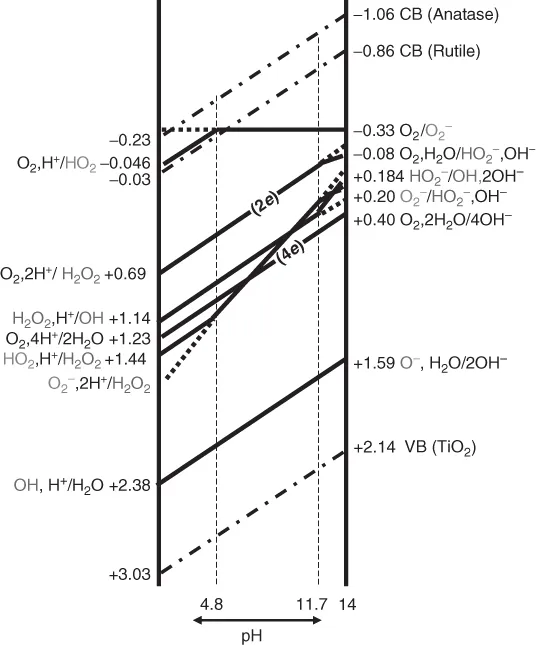
Figure 1.2 shows the standard potentials [8] for the one-electron redox of active oxygen species as a function of pH of the solution. The conduction band (CB) bottom for anatase and rutile TiO2 along with valence band (VB) top of TiO2 is also depicted. The pKa values for H2O2 and OH⋅ are 11.7 and 11.9, respectively [7]. Therefore, the linear lines showing pH dependence in Figure 1.2 change the inclination at the individual pH. It is notable that in the pH range between 10.6 and 12.3, one-electron reduction resulting in OH⋅ formation (Eq. (1.3)) occurs at a higher potential than that resulting in H2O2 formation (Eq. (1.2)). As commonly known, the potential of the VB of TiO2 is low enough to oxidize H2O, suggesting the possibility of the formation of OH⋅. However, the potentials in the figure are depicted based on the free energy change in a homogeneous aqueous solution. Therefore, it does not always mean that the one-electron oxidation of H2O by VB holes at the surface of TiO2 solid takes place in the heterogeneous system. Since the oxidation of H2O to H2O2 and O2 is also possible, only the potential difference between VB and OH⋅ should not be used easily for explaining the possibility of the formation of OH⋅. The competition between OH-radical-mediated reaction versus direct electron transfer has been studied as the effect of fluoride ions on the photocatalytic degradation of phenol in an aqueous TiO2 suspension [9]. Under a helium atmosphere and in the presence of fluoride ions, phenol is significantly degraded, suggesting the occurrence of a photocatalytically induced hydrolysis [9].
Figure 1.2 The standard potentials for the one-electron redox of active oxygen species along with the energy bands of TiO2 as a function of pH of the solution. All redox couples are one-electron process except for those indicated with 2e and 4e.
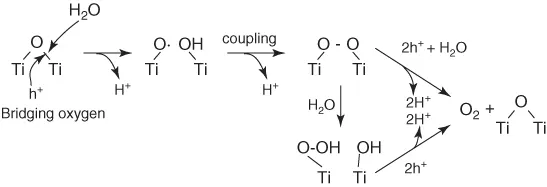
Primary intermediates of water photocatalytic oxidation at the TiO2 in aqueous solution were investigated by in situ multiple internal reflection infrared (MIRIR) absorption combined with the observation of photoluminescence from trapped holes [10]. The reaction is initiated by a nucleophilic attack of a H2O molecule on a photogenerated hole at a surface two hold coordinated O site to form [TiO⋅HO–Ti]. A plausible reaction scheme is shown in Figure 1.3. Detailed investigations revealed the presence of TiOOH and TiOOTi as primary intermediates of the oxygen photoevolution reaction. This means that water is oxidized to form hydrogen peroxide adsorbed on TiO2 surface, but the formation of OH radical in the oxidation process of water was denied.
Figure 1.3 Reaction scheme for the oxygen photoevolution reaction on TiO2 (rutile) in contact with an aqueous solution of pH 1–12.
Ultraviolet photoelectron spectroscopy (UPS) studies showed that the top of the O-2p levels for surface hydroxyl groups (Ti–OH) at the rutile TiO2 (100) face is about 1.8 eV below the top of the VB at the surface [11]. This implies that surface hydroxyl groups cannot be oxidized by photogenerated holes in the VB. On the basis of the electronic structure of surface-bound water obtained from the data reported in the literature of X-ray photoelectron spectroscopy (XPS) study, it is evidenced that water species specifically adsorbed on terminal (surface) Ti atoms cannot be photooxidized under UV illumination [12]. The photogenerated VB free holes are favorably trapped at the terminal oxygen ions of the TiO2 surface (O2−)s to generate terminal (O−)s radicals, rather than being trapped at adsorbed water species to produce adsorbed OH⋅. As discussed later, when OH⋅ is detected in photocatalytic reactions, it should be formed by photocatalytic reduction of H2O2 (Eq. (1.3)).
1.2 Trapped Electron and Hole
Different from the semiconductor bulk, many electronic energy states may be formed within the band gap at the solid surface. These energy levels are capable of trapping VB holes and CB electrons. The trapped energy is considerably larger at the surface than in the bulk, indicating that it is energetically favorable for carriers to travel from the bulk to the surface [13]. At the surface, the trapping sites generally correspond to five-coordinated Ti+ and two-coordinated O− surface ions. When an appropriate acceptor (a scavenger), such as O2 for electrons or methanol for holes, is adsorbed on the surface, it was suggested that the carriers should be preferentially transferred to the adsorbate rather than remain trapped at the surface sites [13].
When there are no molecules that can suffer the reaction, the existence of electrons and holes can be detected at a low temperature such as 77 K. To detect such paramagnetic species, electron spin resonance (ESR) spectroscopy is a valuable method [14, 15].
Holes and electrons could be observed by the absorption spectra just after the short pulse excitation under ambient temperature [16]. Trapped holes show that the absorption peaked at about 500 nm [17] and disappeared by the further reactions. On the other hand, trapped electrons show a broad absorption band that peaked at about 700 nm [18], which react mainly with oxygen molecules in air. Trapped electrons are so stable in the absence of O2 that the kinetics can be explored by means of a stopped flow technique [19]. The reduction kinetics has been investigated through the electron acceptors such as O2, H2O2, and NO3−, which are often present in photocatalytic systems. The experimental results clearly showed that the stored electrons reduce O2 and H2O2 to water by multielectron transfer processes [19]. Moreover, NO3− is reduced via the transfer of eight electrons evidencing the formation of ammonium ions. On the other hand, in the reduction of toxic metal ions, such as Cu(II), two-electron transfer occurs, indicating the reduction of the copper metal ion into its nontoxic metallic form.
1.3 Superoxide Radical and Hydrogen Peroxide (O2⋅− and H2O2)
Si...
Table of contents
Cover
Related Titles
Title Page
Copyright
Editorial Board
Series Editor Preface
Preface
About the Series Editor
About the Volume Editor
List of Contributors
Part I: Fundamentals: Active Species, Mechanisms, Reaction Pathways
Part II: Improving the Photocatalytic Eff icacy
Part III: Effects of Photocatalysis on Natural Organic Matter and Bacteria
Part IV: Modeling. Reactors. Pilot plants
Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Photocatalysis and Water Purification by Pierre Pichat, Max Lu in PDF and/or ePUB format, as well as other popular books in Technology & Engineering & Environmental Management. We have over 1.5 million books available in our catalogue for you to explore.