![]()
Chapter One
Organocopper Chemistry
Bruce H. Lipshutz
Department of Chemistry & Biochemistry
University of California, Santa Barbara, USA
Contents
1. Introduction
2. Click Chemistry: Just Add Copper
3. Cu-Catalyzed Aminations and Amidations
3.1 Aminations
3.2 Amidations
4. Cu-Catalyzed O- and S-Arylations
4.1 Diaryl Ether Formation
4.2 Diaryl Thioether Formation
5. Asymmetric Cu-Catalyzed Borylations
6. Oxidations of Organocopper Complexes
6.1 Biaryl Syntheses
6.2 Aminations
7. 1,2-Additions to Imines
8. Cu-Catalyzed Asymmetric C-C Bond Formation
8.1 Copper Enolates
8.2 Conjugate Additions
8.3 Asymmetric Allylic Alkylations (AAA)
9. Copper Hydride Chemistry
10. Miscellaneous Processes
10.1 C-H Activation
10.2 Protodecarboxylation
10.3 Cu-Catalyzed Carbometallation
11. Conclusions and Outlook
12. Acknowledgments
13. References
1. Introduction
Synthetic Chemistry of Cu(I): Still in Focus and “Hot”
Catalysis. It was the buzzword in the 1990s, and certainly insofar as organocopper chemistry is concerned, there has been no letup on this front in the new millennium. Very good reasons exist for this emphasis, both in terms of development of new methodologies as well as in applications. The key driver is usually the cost of waste disposal; whether copper is contained in an aqueous workup mixture, or in amounts greater than the ppm level allowed in pharmaceuticals, it requires attention. Starting with as little copper as possible in a reaction just makes sense, notwithstanding its “base” metal status. So, whereas the accent in previous versions of the Manual was on reagents, this chapter is organized by reaction type and focuses heavily on processes that are catalytic in copper(I). Nonetheless, attention is also directed to traditional albeit stoichiometric copper chemistry, acknowledging the fundamentals that began with Henry Gilman more than 60 years ago[1] and still are very much valued by the synthetic community today.
Within the catalysis manifold, remarkable advances have been brought to light in both asymmetric as well as achiral synthesis. As already witnessed with precious metal-based technologies, such as asymmetric hydrogenations, the metal is important, but the ligands “rule.” Research over the past decade has led to several mono- and (mostly) bidentate nonracemic ligands that possess, and translate, their extraordinary innate facial biases as their derived copper complexes to a host of substrate types. Several newly discovered species now present themselves to the practitioner, where enantioselectivities are routinely in excess of 90%. On the other hand, chiral upgrades often allow industrial chemists to realize the desired high levels of enantiometric excesses (ee’s) needed in pharma, thereby dettaching any real significance to the “magic” barrier of 90% ee. Nonetheless, the challenges extended by nature to chemists to achieve as close to 100% stereocontrol have not in the past, nor are they likely in the future, to be ignored. Of course, stereocontrol is not the only element that counts in such catalysis; indeed, with up-front costs for copper essentially nonexistent from the perspective of economics, it is turnover number (TON) that may dictate usage. The gap between what academicians may see as “catalytic amounts” (usually 1–5 mol%) and the required low loadings for an industrial process can be hard to fill. Hopefully, some of the progress made as highlighted in this contribution will entice our industrial colleagues.
A wealth of achiral copper chemistry is also covered; in fact, it is still the lion’s share of reports in this field. Whether copper(I) in the form of its salts (CuX), perhaps (in situ) derived organocopper species (RCu), or even a cuprate (R2Cu−M+), there is no denying the textbook status of this metal in organic synthesis. Nonetheless, both silver and especially gold chemistry are making astounding advances. But of the group 11 metals, copper still offers the widest variety of synthetically valued chemoselectivities. And while questions regarding mechanisms, aggregation state(s), and structure of organocopper complexes remain, tremendous progress has been made on these fronts as well. The spirited debate throughout the 1990s on the existence, or not, of “higher order” cuprates provided new incentives for computational, structural, spectroscopic, and physical organic chemistry that has since appeared and has helped to advance the field.
As noted in earlier editions of this Manual, and as is true herein, this chapter is written for the practitioner who faces an ever expanding literature on organometallics. Notwithstanding the emphasis now squarely placed on catalysis, many of the same practical questions arise as highlighted previously, choices of copper salt, solvent, precursor reagent, stoichiometry, and additives, and today, there have been new variables that can play major roles, such as the choice of (e.g., nonracemic) ligand, potential for heterogeneous catalysis, and the option to employ microwave assistance. Thus, with additional insights from several colleagues who have shared their experiences in organocopper chemistry, many of the “secrets to success” are again revealed in this single source. Unfortunately, however, the field is too broad for this opus to be comprehensive; indeed, tough decisions had to be made as to coverage. Thus, there are entire areas even within Cu(I)-catalyzed chemistry that are not included (e.g., cyclopropanations, Diels-Alder constructions, etc.), and surely others that may cause the reader to wonder “What about …?”. The explanation is simple: Each author had a page limitation to his chapter, and it was recommended (mainly as a result of technical matters associated with binding) that the editor not violate his own rules!
2. Click Chemistry: Just Add Copper
Reviews. Diez-Gonzalez, S. Curr. Org. Chem. 2011, 15, 2830; Diez-Gonzalez, S. Catal. Sci Technol. 2011, 1, 166; Cantillo, D.; Avalos, M.; Babiano, R.; Cintas, P.; Jimenez, J. L.; Palacios, J. C. Org. Biomol. Chem. 2011, 9, 2952; Elchinger, P-H.; Faugeras, P-A.; Boens, B.; Brouillette, F.; Montplaisir, D.; Zerrouki, R.; Lucas, R. Polymers, 2011, 3, 1607; Becer, C. R.; Hoogenboom, R.; Schubert, U. S. Angew. Chem., Int. Ed. 2009, 48, 4900 (metal-free); Meldal, M.; Tornoe, C. W. Chem. Rev. 2008, 108, 2952; Lutz, J.-F. Angew. Chem., Int. Ed. 2007, 46, 1018; Fokin, V. V.; Wu, P. Aldrichimica Acta 2007, 40, 7; Bock, B. D.; Hiemstra, H.; van Maarseveen, J. H. Eur. J. Org. Chem. 2006, 51.
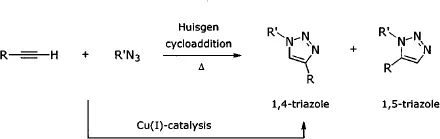
The 2002 papers by the groups of Sharpless[2] at Scripps (La Jolla, CA) and Meldal[3] at the Carlsberg Laboratory (Denmark) highlighting the remarkable acceleration of Huisgen cycloadditions between organic azides[4] and terminal alkynes by Cu(I) have led to an avalanche of renewed interest in, and usage of, copper(I) in synthesis. The facility with which these two relatively high-energy educts “click” to form heteroaromatic 1,2,3-triazoles is truly impressive, and the community at large is
using this chemistry in both routine and highly innovative ways. Importantly, these otherwise thermally driven cycloadditions, which often lead to mixtures of 1,4- and 1,5-disubstituted triazoles,[5] are fully controlled in the presence of Cu(I) to afford only the 1,4-regioisomer[6] (while Ru leads to the corresponding 1,5-isomer).[7] Mechanistic details have not been fully elucidated, but significant progress has been made, most notably by Finn[8a] and Fokin,[8b] and more recently using density functional theory (DFT) calculations.[9] The data suggest involvement of copper(I) acetylides, shown to be associated within a dimeric array. Electron-withdrawing groups on the alkyne increase reactivity. A key point for the s...