![]() Part I Research and Development
Part I Research and Development![]()
1
Discovery of New Medicines
Yves J. Ribeill
Scynexis Inc., Research Triangle Park, NC, USA
Introduction
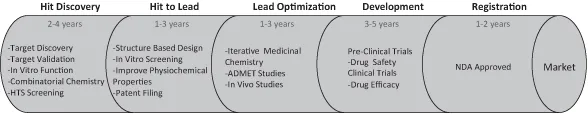
Patients rely on discovery researchers to embrace innovation, make advances and deliver new therapies that will improve their lives. The discovery of drugs is a complex, costly and lengthy process involving several distinct stages on the path towards deliveryof a marketable drug (Figure 1.1). It is becoming increasingly important in the ever competitive enterprise of drug discovery for researchers to develop innovative drug discovery strategies in order to fill their pipelines. This chapter is designed to highlight these modern approaches to drug discovery and the changing therapeutic landscape for the currently available drugs.
Progress in drug discovery relies on fundamental biological research in pharmaceutical and biotechnology companies as well as academia to identify new biological targets, to implement target validation strategies and to confirm target relevance in a disease state. Initially, drug discovery researchers select a target that can interact with a modulator, such as a protein or small molecule. After a target has been chosen, researchers must demonstrate that the target is relevant to a disease in both living cells and animal models. The promise of determining the whole genome sequence, new insights into molecular sources of disease, technological advances in both target and lead validation, and high-throughput screening (HTS) strategies all provide potentially novel opportunities for target validation in drug discovery. After such validation, the search begins for a ‘hit’ molecule that interacts with the desired target. These ‘hits’ may originate from nature, de novo design or HTS but, in most cases, require optimisation to ‘leads’ via cycles of altering the structure and properties of the molecules followed by iterative screening. During this process, lead compounds are further optimised for the desired absorption, distribution, metabolism, excretion and toxicological (ADMET) properties. ADMET optimisation supplies the ‘lead compound’, which advances to later stages of drug development.
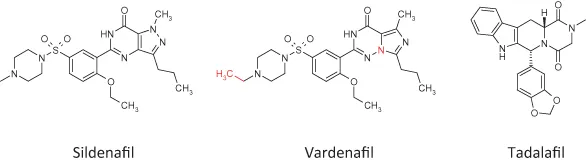
A case study around the investigation of phosphodiesterase (PDE) inhibitors illustrates the successful applications of the principles of contemporary drug discovery and development. Based on the discovery of an endothelium-derived relaxing factor and the interplay of nitric oxide, cyclic guanosine monophosphate (cGMP) and PDEs in vasodilation, researchers at Pfizer reasoned that a PDE inhibitor might be advanced for the treatment of angina [1]. Their comparison of the structure of cGMP, with consideration of how it may bind to PDEs, with that of the weak vasodilator Zaprinast, also a PDE inhibitor, further supported their hypothesis. The screening of existing compound collections as well as the rational design of analogues produced active molecules that targeted PDE-5, a cGMP-specific PDE located in coronary smooth muscle. Further optimisation of these compounds for the desired potency and ADMET properties led to a clinical candidate for angina; the compound was found to be ineffective, and its development as a cardiovascular drug was halted. During the clinical trials, however, some patients reported experiencing enhanced penile erections. Subsequently, PDE-5 was identified as the main cGMP-degrading enzyme in the corpus cavernosum. Thus, efforts were redirected toward proving the effectiveness of the lead compound as a treatment for erectile dysfunction and the eventual approval of sildenafil (Viagra) in 1998 as a prescription medicine for erectile dysfunction [2,3].
Important parts of drug discovery and development are intellectual property protection and the ability to navigate around prior art. Pfizer filed patent applications proactively around the lead compound/series, as well as its therapeutic use, to deter competitors from achieving success in the PDE arena in similar chemical space. Others interested in advancing compounds in this therapeutic area became faced with searching for gaps in the patent coverage or pursuing alternative structural classes. Implementation of a ‘patent busting’ strategy enabled the discovery of vardenafil (Levitra). Analogues outside of the Pfizer patents were identified, optimised and evaluated in clinical trials ahead of product launch in 2003. Conversely, Icos and Lilly investigated an unrelated molecule as a longer-acting PDE-5 inhibitor that led to the approval of tadalafil (Cialis), also in 2003. Figure 1.2 highlights the structural similarities, and differences, of these three medicines.
Medicines Marketed in the Years 2008–2011
The pharmaceutical and biotechnology industries spend billions on cutting-edge research and development (R&D), clinical trials and marketing to introduce drugs to market. However, fewer than one in 50 drug discovery projects results in the delivery of a drug to market [4], and the average time from concept to market is 15 years, at a cost of nearly a billion dollars per drug [5]. Further, since 2008, new drug approvals have declined sharply despite an increase in R&D spending [6]. The observed high attrition rate is unsustainable and researchers must constantly reassess their tactics in order to translate discovery research into clinical success.
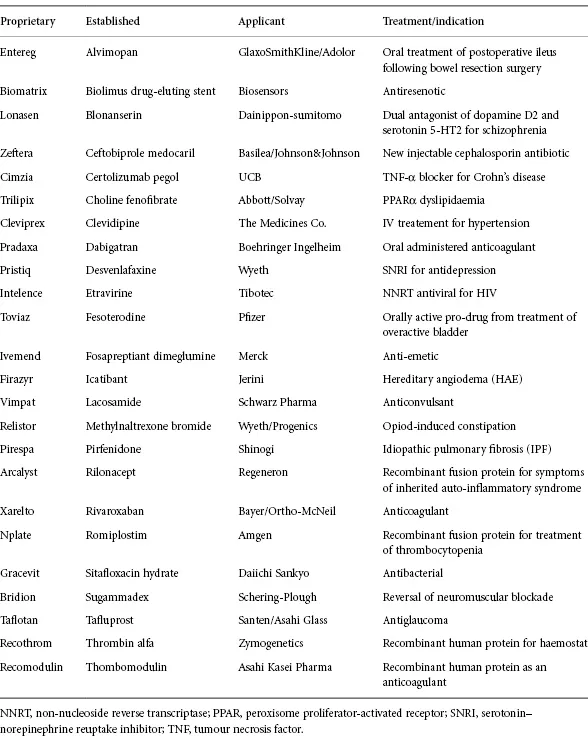
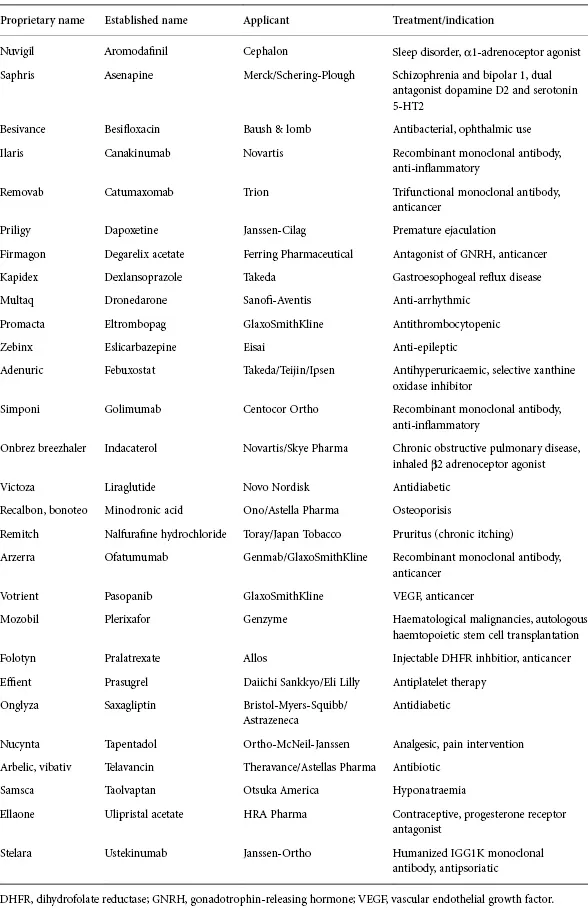
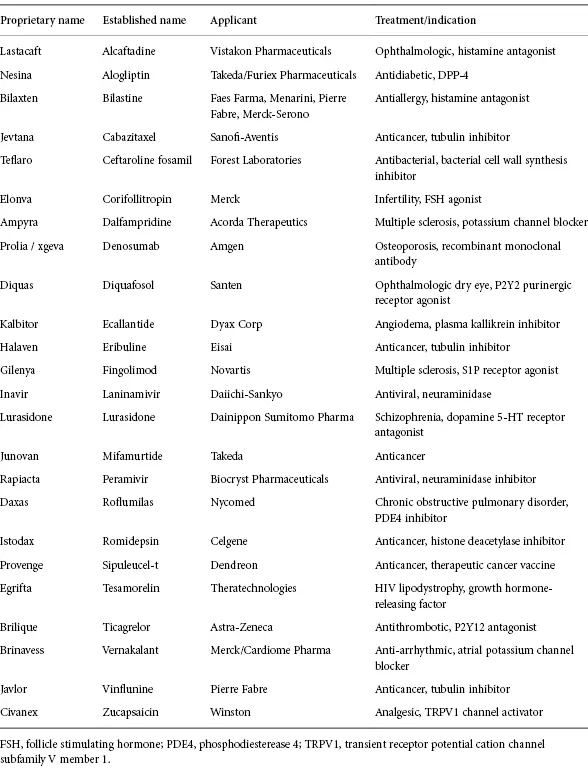
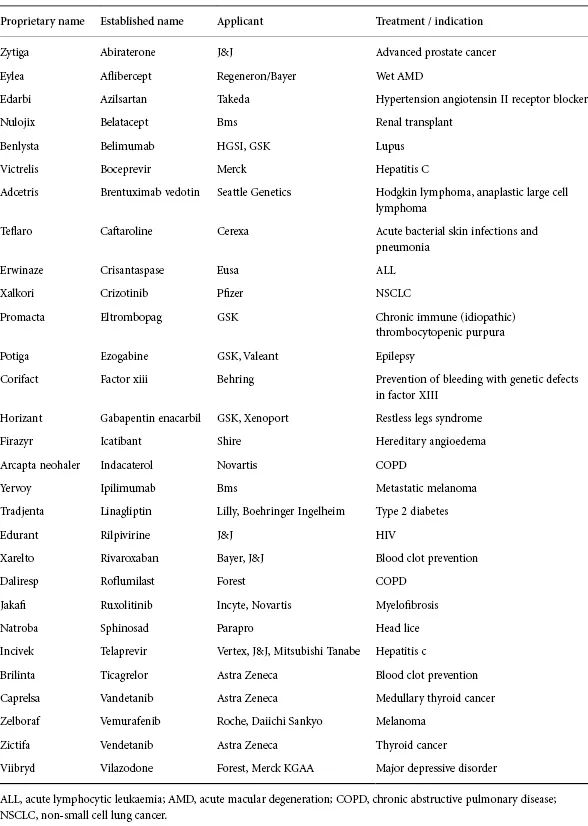
Despite the steady decline in overall new drug approvals there has been a steady increase of new products in the therapeutic areas of anti-infective, metabolic and orphan diseases, as well as a shift into specialty-care therapies (Tables 1.1, Table 1.2, Table 1.3 and Table 1.4) [7–9]. The majority of new molecular entities (NMEs) continue to be small molecules; however, vaccines and non-biological oligonucleotides employed as macromolecular therapeutics are directed at enzymes and receptors that have been classically modulated by small molecule drugs.
Table 1.1 New molecular entities (NME) approved 2008.
Reproduced from Hegde S, Schmidt M. To Market, To Market Annual Reports in Medicinal Chemistry 2009; 44: 577 with permission from Elsevier [8]
Table 1.2 NME approved 2009.
Reproduced from Hegde S, Schmidt M. To Market, To Market Annual Reports in Medicinal Chemistry 2010; 45: 467 with permission from Elsevier [9]
Table 1.3 NME approved 2010.
Reproduced from Bronson J, Dhar M, Ewing W, Lonberg N. To Market, To Market Annual Reports in Medicinal Chemistry 2011; 46: 433 with permission from Elsevier [7]
Table 1.4 NME approved 2011.
Reproduced with permission from U.S. Food and Drug Adminstration, ‘How Drugs are Developed and Approved?’, http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/DrugandBiologicApprovalReports/ucm121136.htm. Last accessed 13 Aug 2012
In response to the decline in new drug approvals, new approaches have been put in place:
1. drug combinations that target multiple pathways continue to increase in drug discovery to modulate the interplay of complex chemical pathways involved in diseases;
2. drug repurposing has accounted for two-thirds of new drug applications. Increased focus on repurposing existing drugs for orphan indications emanates from disease-focused philanthropic groups; and
3. collaboration strategies between pharmaceutical companies and academic research institutions have contributed to the drug discovery process [10,11].
While academic research is focused principally on the underlying mechanistic components of a disease and the pharmaceutical industry focuses on progressing discovery projects, a willingness to share expertise through these research alliances has resulted in advances in poorly funded rare diseases.
Impact of High Throughput Screening in Drug Discovery
A judicious choice of therapeutic area and biological target, along with an acceleration of development time through scientific innovation, are critical to a successful R&D drug discovery programme. Pharmaceutical companies often engage in economic balancing when choosing therapeutic areas in which to begin research by weighing the probability of delivering a product against potential sales of this product. Additionally, companies must consider development costs and regulatory hurdles when choosing their research path. As an example, since 2000, the proportion of R&D projects from available corporate portfolios in the area of antineoplastic agents has increased by about 7% [12]. The average sales per year for an antineoplastic agent developed since 2000 was 92 million dollars, among the highest of the major therapeutic classes. However, the probability of success for reaching the market from the preclinical phase for an antineoplastic development project is quite low due to project attrition [12].
The drug discovery paradigm has evolved in recent times. In the simplest example, the mode of action of a compound (drug) centres on its binding to a receptor (target) that influences a biochemical pathway, which is relevant to a physiological process, and the sum of these events provides a therapeutic benefit to a disease state. In many cases the reality is not that simple, thus additional approaches have become necessary. Prior to 1990, the standard approach to small molecule drug discovery relied on iterative, low-throughput in vivo screening and optimisation of compounds to improve chemical or biochemical parameters (e.g. potency, selectivity or pharmacokinetic properties). Antihypertensive beta-blockers were developed through this process from adrenaline (epinephrine) [12] where analogues were synthesised individually and evaluated in concurrent assays (often in vivo) and optimised via medicinal chemistry to progress compounds to clinical trials. With the advent of HTS and the availability of large collections of compound libraries, this model was criticised for being slow, expensive and obsolete. Target selection has since become heavily influenced by the compatibility of that target with an HTS approach to enable rapid and cost-effective evaluation of hundreds of thousands of compounds from screening collections.
Much debate continues around the relative effectiveness of the two models outlined earlier. The premise that high-affinity binding to a single biological target that is associated with a disease state will afford a therapeutic benefit in humans [13,14] has been countered by opinions that pharmaceutical products developed pre-HTS in fact do not act on a single target, and are actually more promiscuous than previously thought, thereby exposing a deficiency in the HTS approach. Off-target binding could have an important role in the efficacy of a drug candidate that: (i) underscores not only the importance of target selection, but also the choice of the R&D strategic model; and, (ii) provides one plausible explanation for the success of the in vivo screening model in delivering NMEs.
Impact of Combinatorial Chemistry on Drug Discovery
The paradigm of drug discovery experienced changes at the end of the last century. The acceptance of HTS methodologies reinforced opinions to prepare larger, diverse collections of test compounds, especially peptides and small molecules. Solid-phase chemistry [15] enabled the assembly of complex polypeptides on a polymer support, simultaneously providing access to previously unattainable molecules and foreshadowing the use of automation. The overarching features of this approach were the use of substrates covalently bound to a solid-support (polypropylene, polystyrene or other polymeric bead...