Demonstrating how and why to measure physicochemical and biomimetic properties in early stages of drug discovery for lead optimization, Physicochemical and Biomimetic Properties in Drug Discovery encourages readers to discover relationships between various measurements and develop a sense of interdisciplinary thinking that will add to new research in drug discovery. This practical guide includes detailed descriptions of state-of-the-art chromatographic techniques and uses real-life examples and models to help medicinal chemists and scientists and advanced graduate students apply measurement data for optimal drug discovery.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
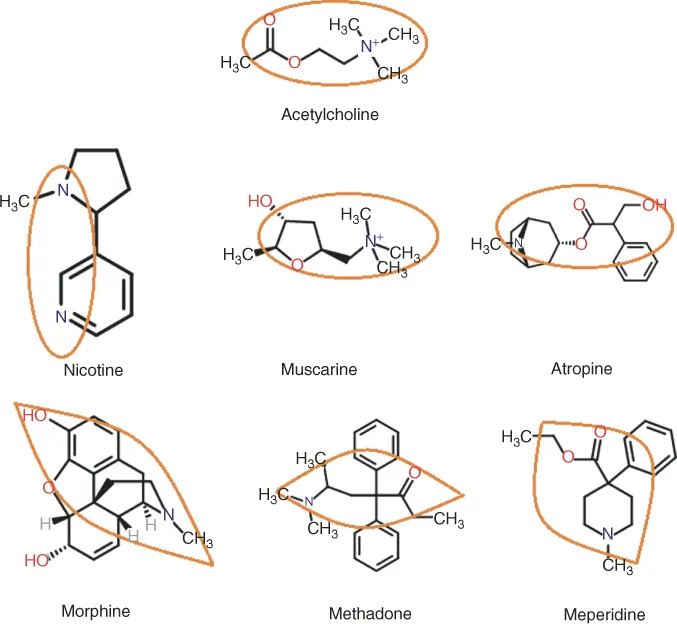
The way we discover drugs now is very different from the process followed 30–50 years ago [1–3]. In the past, the drug discovery process was based on the structure of known active compounds. The structure of the active compound, which was either endogenous (e.g., acetylcholine, adrenaline, and steroid hormones) or a natural product (e.g., morphine, papaverine, cocaine, atropine, and digitalis glycosides), was identified and modified by a trial-and-error method. Scientists isolated the active material, revealed the structure and the mechanism of the pharmacological action, and based on this knowledge they would modify the chemical structures with the aim of improving the activity, decreasing the toxicity, and increasing the duration of action. Using this approach, they discovered, for example, a series of cholinesterase inhibitors, a series of adrenaline analogs for sympatomimetic or sympatolytic compounds, semisynthetic steroid hormones, and many more. Another example is pethidine containing the pharmacophores of morphine. The ethoxy derivative of papaverine called No-Spa has a longer half-life and is a stronger spasmolytic agent. Figure 1.1 shows a few analog-based drug molecules designed from natural compounds. There are numerous examples of naturally occurring molecules or their derivatives being used as drugs, such as warfarin (anticoagulant derived from dicumarol found in sweet clover, hirudin (anticoagulant from leeches), statins (a fungal metabolite that reduces plasma cholesterol), vinca alkaloids (anticancer drugs isolated from periwinkle plant families), and antibiotics (derived from fungal metabolites).
Today, drug discovery utilizes recent advances in biochemistry and genetics. Drug discovery usually starts with discovering a so-called “target” that can be enzymes or receptors, such as G protein-coupled receptors (GPCR) and enzyme targets, such as kinases, ion channels, and hormone receptors.
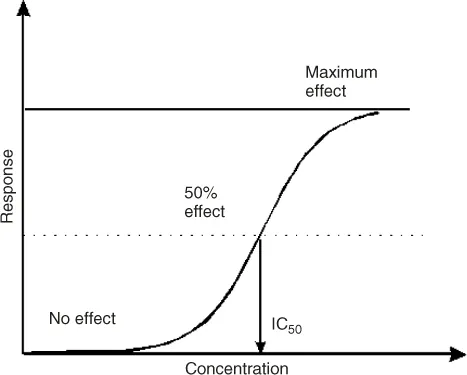
The medicinal chemists and computational chemists work closely together to generate ideas about potential active molecules that would fit to the “target” molecule. They develop a so-called high throughput screening (HTS) method that enables the big pharmaceutical companies to screen large number of compounds against a particular target. Compounds that show activity in HTS are called hits. The activity of the “hits” is further evaluated by repeated experiments for obtaining the standard concentration–potency curve. Figure 1.2 shows a typical concentration–response/potency profile during hit generation and confirmation.
Figure 1.1 Drug molecules designed as analogs of natural active molecules containing the active pharmacophore.
Figure 1.2 Typical concentration versus potency profile used to derive the pIC50 values that refer to a concentration of the compound producing 50% of the maximum measurable response in a particular screen.
This phase of the drug discovery process is often called hit generation. Synthetic chemists then synthesize a whole library of molecules, making numerous combinations of substituents on an active “skeleton” or scaffold. This part of the process is often called lead generation. The “lead” molecule usually shows high activity on a particular target. It is advisable to test the selectivity of the lead molecule to avoid so-called promiscuous binders that produce a hit on a variety of targets; that is, the lead molecule should show activity preferably only on one target. The “lead” molecule also should show reasonable solubility and bioavailability during the first in vivo animal experiments.
Sometimes it is possible to get the three-dimensional structure of the targets using X-ray crystallography or nuclear magnetic resonance (NMR) and the binding sites are identified. Computational chemists, using various docking methods, find or design small molecules that fit to the active site of the target [4]. The primary role of a docking program is to identify biologically active compounds that may act as ligands to a target binding site based on the calculated intermolecular interaction energy. When the X-ray crystallographic structure is not available, there are various computational methods that can be used to assess the binding site of the target protein and design molecules that will bind to it.
The drug discovery process moves into the lead optimization phase, where molecules then undergo a series of structural modifications, which helps to establish a so-called SAR (structure–activity relationship) for the series. It allows the chemists to establish which part of the molecule is essential for activity. During this process, they often find much more active analogs than the original lead molecule. The activity measurements are repeated using cell-based assays or in the presence of plasma proteins. Usually that is the point where potential liabilities may be observed. The reduced cellular activity could be due to low permeability or solubility or strong nonspecific binding to cellular components. This is the time when they approach the physicochemists to measure solubility, permeability, and protein binding in order to determine the cause of the loss of cellular activity. Usually the activity is not lost completely but only weakens. This is called drop off of the activity. The lead optimization process often takes a year or two, maybe even considerably longer. It includes various in vitro assays for the assessment of selectivity and in vitro microsomal stability. For a smaller number of potentially promising compounds, animal studies, bioavailability measurements, and pharmacokinetic studies are also carried out before candidate selection. At this stage, scientists try to establish the dose necessary for effective in vivo pharmacological activity. It is important to observe in vivo efficacy and receptor or target engagement assays to prove that the observed pharmacological activity is a consequence of the molecule binding to the target. The first toxicological studies are also performed at this stage.
Chemists utilize information from other disciplines to establish structure–property relationships and modify the structures accordingly. If successful, at the end of this process, the molecule that shows the desired potency, bioavailability, in vivo receptor occupancy, and acceptable pharmacokinetic properties will be declared as the “candidate” and will then enter a long development process. When a molecule is identified as a candidate, the research process ends and the selected molecule is passed over to development scientists, who then carry out preclinical studies, extensive toxicity studies using much higher doses than are needed for efficacy, formulation studies, large-scale synthesis, and preparation for the first clinical trial “first time in human.” Usually, a “backup” compound is also selected in case the candidate molecule fails during the development process.
During this process, we also need a disease model to prove the concept that the compound acting on the selected target actually can cure the disease. It is called proof of concept and target validation. Figure 1.3 summarizes the major phases of the early drug discovery process. The high throughput chemistry is often replaced nowadays by lower throughput synthesis of smaller number of compounds as a designed library. The HTS campaign is very expensive; therefore, it is used when hit molecules are not available from other sources.
Figure 1.3 Major steps of the modern drug discovery process including both the biological and chemical aspects.
However, traditional HTS has not delivered on its promise of increasing the numbers and quality of new drugs entering clinical trials. The typical hit rate of a screening campaign is usually less than 1%. This lack of success is due in part to the complexity and the relatively large size of the compounds routinely being screened. On the basis of the mathematical probability, the ability of finding perfect binding, including four to six interactions at the binding site, is very low [5].
Recently, the so-called fragment approach has been used more often. It has been found that the probability of binding decreases rapidly as the complexity of the ligand increases. However, there is much higher probability of finding one or two binding interactions at a time and then mapping the other possible interactions at the binding site one by one. Therefore, instead of screening a very large number of molecules to find the one that shows affinity to the receptor, there is much higher probability of success in finding molecular fragments that have only one or two binding interactions. From the small, so-called “fragment” molecules, we can build bigger molecules by combining several fragments into a single molecule. In this way, we can increase the likelihood of finding a highly potent larger molecule. The effective use of the fragment-based approach is dependent on the use of biophysical techniques such as X-ray crystallography or NMR measurements to identify how and where the fragment binds to the target. Traditional bioassays used in HTS are generally unable to detect such small drug fragments because of their low potency binding to the protein target. The approach requires screening of molecules at higher concentration (millimolar and not micromolar or nanomolar) to be able to detect the weaker interactions. The fragment-based drug design approaches are adopted by many big and small pharmaceutical companies. The typical hit rate is usually an order of magnitude higher than the HTS hit rate (around 10%). The typical binding interactions between such small molecules and the target are hydrogen bonding (by donating or accepting proton), hydrophobic, and
interactions. The small molecule has to fit spatially well to the binding sites, so that the interacting molecular forces can produce energetically stable binding. Ideally, fragment molecules should allow chemists to attach other functional groups and grow the molecules. It means that the fragments should be chemically tractable for modification.
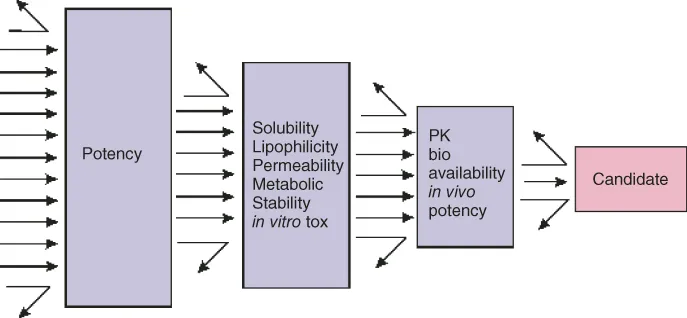
The early drug discovery process sometimes produces several thousands of potent molecules, still only a few reach the market. The attrition is very high. Although this part of the drug discovery research is expensive, it costs only around 5% of the total cost of getting a drug molecule to the market, which is estimated to be around $800 million. Unfortunately, only 1 in 10 candidate molecules obtain approval from the Food and Drug Administration (FDA). This huge attrition of the compounds is due to so-called “developability” issues. If a candidate is to fail, the earlier the better, as it saves a lot of effort and investment for the company. The most common reasons for stopping the candidates from further development are disadvantageous pharmacokinetics, lack of efficacy in the clinic, toxicity, and formulation problems. Sometimes development is stopped for commercial reasons, as other compounds by competitors appear on the market. The potential disadvantageous pharmacokinetics includes poor oral exposure, high clearance, drug–drug interaction, too short or too long half-life, etc. The physicochemical properties of the molecules have a crucial impact on these failures [6–8]; therefore, it is vital to include these measurements early in the drug discovery process. Figure 1.4 shows how the developability considerations and physicochemical properties are built into the candidate selection process.
Figure 1.4 Developability considerations and physicochemical properties should be taken early in the drug discovery process.
For example, poor solubility of the discovery compounds may compromise the results of the screening. It may produce false-positive or false-negative potency values. Very lipophilic compounds often bind to many different types of proteins and to various nonspecific binding sites as well, thus reducing the actual available free concentration of the compound and shifting the pIC50 values. Ionized compounds can have various ionic interactions; they ...
Table of contents
Cover
Title Page
Copyright
Preface
Chapter 1: The Drug Discovery Process
Chapter 2: Drug-Likeness and Physicochemical Property Space of known Drugs
Chapter 3: Basic Pharmacokinetic Properties
Chapter 4: Principles and Methods of Chromatography for the Application of Property Measurements
Chapter 5: Molecular Physicochemical Properties that Influence Absorption and Distribution—Lipophilicity
Chapter 6: Molecular Physicochemical Properties that Influence Absorption and Distribution—Solubility
Chapter 7: Molecular Physicochemical Properties that Influence Absorption and Distribution—Permeability
Chapter 8: Molecular Physicochemical Properties that Influence Absorption and Distribution—Acid Dissociation Constant—pKa
Chapter 9: Models with Measured Physicochemical and Biomimetic Chromatographic Descriptors—Absorption
Chapter 10: Models with Measured Physicochemical and Biomimetic Chromatographic Descriptors—Distribution
Chapter 11: Models with Measured Physicochemical and Biomimetic Chromatographic Descriptors—Drug Efficiency
Chapter 12: Applications and Examples in Drug Discovery
Appendix A: Answers to the Questions for Review
Appendix B: List of Abbreviations and Symbols
Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Physicochemical and Biomimetic Properties in Drug Discovery by Klara Valko in PDF and/or ePUB format, as well as other popular books in Medicine & Pharmacology. We have over 1.5 million books available in our catalogue for you to explore.