
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Modern Drug Synthesis
About this book
Following Contemporary Drug Synthesis and The Art of Drug Synthesis (Wiley, 2004 and 2007), two well-received works, is this new book that demystifies the process of modern drug discovery for practitioners and students. An enhanced introduction covers areas such as background, pharmacology, SAR, PK/PD, efficacy, and safety. Focusing on the advantages of process synthesis versus the discovery synthetic route, Modern Drug Synthesis features authoritative coverage by distinguished editors and authors (some chapter authors are the actual inventor of the drug) of twenty different drug molecules.
Tools to learn more effectively

Saving Books

Keyword Search

Annotating Text

Listen to it instead
Information
PART I
INFECTIOUS DISEASES
Chapter 1
Raltegravir (Isentress): The First-in-Class HIV-1 Integrase Inhibitor
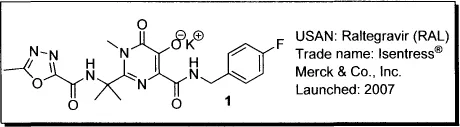
1.1 Background
HIV/AIDS is a global pandemic. Nearly three decades of HIV/AIDS research has resulted in the development of more than 20 antiretroviral drugs approved by the U.S. Food and Drug Administration (FDA) for treatment of the disease; combinations of these drugs, known as highly active antiretroviral therapies (HAART), have dramatically decreased morbidity and increased life expectancy.1 Nevertheless, HIV/AIDS remains a significant cause of morbidity and mortality worldwide. The Joint United Nations Program on HIV/AIDS estimated in 2007 that more than 33 million people lived with the disease worldwide and that AIDS killed more than 2 million people, including 330,000 children. In the United States alone, the Centers for Disease Control and Prevention (CDC) estimated in 2006 that more than one million people were living with HIV infection.2–4 Although antiretroviral drugs have undeniably changed the lives of many HIV-positive individuals, an unmet medical need clearly exists.
Raltegravir, or Isentress (1), is the first FDA-approved inhibitor of HIV integrase. HIV/AIDS drugs are categorized according to their mode of action as nucleoside and nucleotide reverse transcriptase inhibitors [NRTIs, e.g., tenofovir (2)], nonnucleotide reverse transcriptase inhibitors [NNRTIs, e.g., efavirenz (3)] protease inhibitors [PIs, e.g., ritonavir (4)], fusion inhibitors [e.g., enfuvirtide (5)], entry inhibitors [e.g., the CCR5 antagonist maraviroc (6)], and integrase inhibitors (INSTIs, e.g., raltegravir).
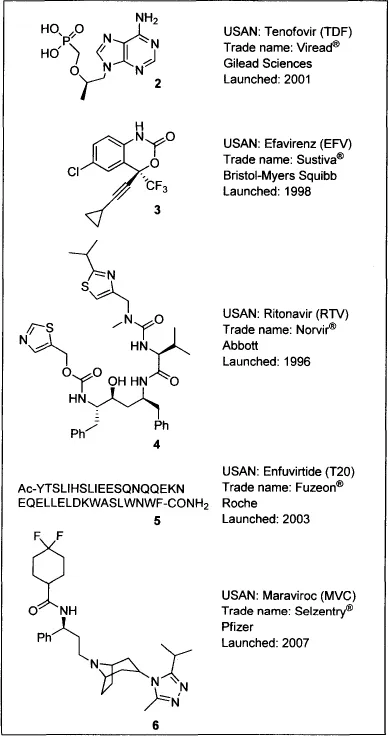
Until recently, all FDA-approved drugs for HIV/AIDS treatment targeted either the viral reverse transcriptase (RT) or protease (PR) enzymes, and treatment guidelines specified that HAART multidrug cocktails should comprise one NNRTI or one PI in combination with two NRTIs.5 Two of the most significant limitations on the effectiveness of these HAART combinations have been drug-related toxicities and the emergence of resistant viruses.6 Drugs with novel modes of action—such as viral entry inhibitors and integrase (IN) inhibitors—have been sought to offer antiretroviral treatment-experienced HIV patients who harbor a drug-resistant virus or suffer toxicities with HAART an opportunity to achieve immunologic recovery and virologic suppression. IN inhibitors have been of particular interest to HIV/AIDS researchers because, unlike RT and PR, IN has no human homolog, and thus inhibitors of IN might be better tolerated at high doses.7
Raltegravir (1) is the first commercially available antiretroviral agent to target IN; at present, it is the only IN inhibitor approved for clinical use. Launched in 2007, raltegravir was originally indicated for combination therapy with other antiretroviral agents in treatment-experienced adults with evidence of viral replication and multidrug-resistant HIV-1 strains. In July 2009, the FDA approved an expanded indication for raltegravir to include treatment-naive adult patients, and in December 2009, the U.S. Department of Health and Human Services (DHHS) revised its HIV treatment guidelines to add a raltegravir combination to the preferred regimens for treatment-naive HIV patients.5 In this chapter, the pharmacological profile and chemical syntheses of raltegravir will be described in detail.
1.2 Pharmacology
The HIV-1 replication cycle involves three key viral enzymes, all of which represent targets for antiretroviral drugs: RT, PR, and IN. RT and PR inhibitors are well represented in the HIV/AIDS treatment armamentarium, but until recently, inhibition of IN had not been successfully exploited in the clinic, despite nearly 20 years of intensive research.8
The enzymatic mechanisms of IN have been extensively reviewed.9 IN catalyzes the insertion of viral genetic material into human DNA through a multistep process that includes 3’-processing, (removal of the terminal dinucleotide from each 3’-end of the viral DNA) and strand transfer (joining of the viral DNA to the host DNA). Both 3’-processing and strand transfer are catalyzed by a triad of acidic residues, D64, D116, and E152, that bind divalent metals such as Mg2+. Divalent metals are required for 3’-processing and strand transfer and also for the assembly of the preintegration complex, which allows viral genetic material to cross the host nuclear membrane and access the host genome.
Raltegravir (1), like its precursors the 1,3-diketo acids (see Section 1.3), inhibits the strand-transfer activity of IN.10 The IN-inhbitory activity of raltegravir is most likely the result of functional impairment of the active site arising from chelation of the critical divalent metal cofactors.11 Raltegravir is both a potent (IC50 = 10 nM) and a highly selective inhibitor of integrase activity, found to be essentially inactive (IC50 > 50 μM) against related enzymes such as hepatitis C virus (HCV) polymerase; HIV RT; HIV RNase-H; and human α-, β-, and γ-polymerases. In a multiple-cycle cell-based assay (human T-lymphoid cells infected with a laboratory strain of HIV-1 in 50% human serum), raltegravir effectively inhibited (IC95 = 31 nM) the replication of HIV-1 infection.12
1.3 Structure–Activity Relationship (SAR)
The 4-aryl-2,4-diketobutanoic acid class of IN inhibitors (also known as 1,3-diketo acids, or DKAs) was discovered independently by researchers from Merck and Shionogi, with patents from both groups published in the same year.13 From a random screen of > 250,000 compounds, the Merck group identified DKAs as the most active IN inhibitors. Compound 7 was the most potent compound found in this screen (Table 1), completely inhibiting HIV-1 infection in a cell-based assay at a concentration of 10 μM.10
Table 1. Activity of DKAs and Related Structures against HIV-1 Integrase
| Entry | Compound | HIV-1 Strand Transfer IC50 (μM) |
| 1 | 1 | 0.01 |
| 2 | 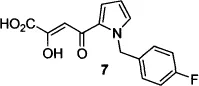 | 0.08 |
| 3 | 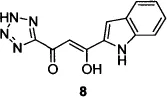 | 0.65 |
| 4 | 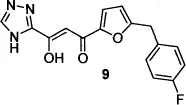 | 0.02 |
| 5 | 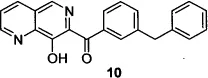 | 0.04 |
| 6 | 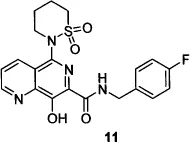 | 0.01 |
| 7 | 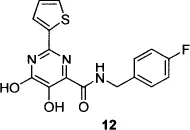 | 0.08 |
| 8 | 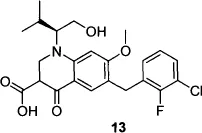 | 0.007 |
The Shionogi group’s first patent13d described compounds (e.g., 8) in which an isosteric tetrazole replaced the carboxylic acid group in the critical but biologically labile DKA pharmacophore. Compound 8, also known as 5CITEP, inhibited HIV-1 3’-processing as well as strand-transfer activity,14 and was the first inhibitor co-crystallized with IN.15 A subsequent patent from the Shionogi group described the systematic modification of the 5CITEP framework to include a variety of heterocyclic replacements for the indole and tetrazole moieties, culminating in the first clinically tested HIV-1 IN inhibitor, compound 9, also known as S-1360.16,17 Clinical development of S-1360, undertaken by a Shionogi–GSK joint venture, was halted when the compound failed in efficacy studies in humans (due to formation of an inactive metabolite via reduction at the carbon adjacent to the triazole).18
The Merck group’s efforts to find a more stable substitute for the DKA pharmacophore resulted in the design of 8-hydroxy-[1,6]naphthyridines such as compound 10,19 wherein the keto-enol-acid triad was replaced with a 1,6-naphthyridine ketone bearing a phenolic hydroxyl group. Further refinement of compound 10—replacement of the naphthyridine phenyl ketone with a 4-fluorobenzyl carboxamide and addition of a six-membered sulfonamide at the 5-position of the naphthyridine core—resulted in compound 11, the second IN inhibitor to reach the clinic.20 The discovery of liver toxicity in long-term safety studies of compound 11 in dogs led to the suspension of clinical development21 of this compound.
Concurrent with the Merck group’s efforts to find an IN inhibitor that would be successful in the clinic, a separate Merck group working on inhibitors of HCV polymerase discovered that dihydroxypyrimidine carboxamide 12 (which, like the other compounds shown in Table 1, evolved from DKA lead structures) was a potent inhibitor of HIV-1 strand transfer, but completely inactive against HCV polymerase.22 Modification of the dihydroxypyrimidine core to the corresponding N-methylpyrimidinone23 followed by optimization of the N-methylpyrimidinone series with respect to metabolic stability, pharmacokinetic profile, antiviral activity, and genotoxicity led to the identification of raltegravir, the first IN inhibitor to be approved by the FDA.12
A second IN inhibitor has reached Phase III clinical trials since the launch of raltegravir. Elvitegravir (13) is a dihydroquinolone carboxylic acid; the monoketo acid motif of this series of inhibitors is proposed to mimic the keto-enol-acid triad of the DKA lead structures.24 Like raltegravir, elvitegravir is a specific inhibitor of HIV-1 strand transfer.25
1.4 Pharmacokinetics and Drug Metabolism
The plasma half-life of raltegravir (1) in rats was 7.5 h and in dogs was 13 h. Plasma half-life was biphasic in both species, ...
Table of contents
- Cover
- Half Title page
- Title page
- Copyright page
- Preface
- Contributors
- Part I: Infectious Diseases
- Part II: Cancer
- Part III: Cardiovascular and Metabolic Diseases
- Part IV: Central Nervous System Diseases
- Part V: Miscellaneous
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Modern Drug Synthesis by Jie Jack Li, Douglas S. Johnson, Jie Jack Li,Douglas S. Johnson in PDF and/or ePUB format, as well as other popular books in Scienze fisiche & Chimica organica. We have over one million books available in our catalogue for you to explore.