![]()
1
Overview of Risk-Based Approach to Phase Appropriate Validation and Instrument Qualification
CHUNG CHOW CHAN
CCC Consulting
HERMAN LAM
Wild Crane Horizon Inc.
XUE MING ZHANG
Apotex, Inc.
STEPHAN JANSEN, PAUL LARSON, CHARLES T. MANFREDI, AND WILLIAM H. WILSON
Agilent Technologies Inc.
WOLFGANG WINTER
Matthias Hohner AG
1 Risk-Based Approach to Pharmaceutical Development
In the United States, the U.S. Food and Drug Administration (FDA) ensures the quality of drug products using a two-pronged approach involving review of information submitted in applications as well as inspection of manufacturing facilities for conformance to requirements for current good manufacturing practice (cGMP). In 2002, the FDA, together with the global community, implemented a new initiative, “Pharmaceutical Quality for the 21st Century: A Risk-Based Approach” to evaluate and update current programs based on the following goals:
- The most up-to-date concepts of risk management and quality system approaches are incorporated while continuing to ensure product quality.
- The latest scientific advances in pharmaceutical manufacturing and technology are encouraged.
- The submission review program and the inspection program operate in a coordinated and synergistic manner.
- Regulatory and manufacturing standards are applied consistently.
- FDA resources are used most effectively and efficiently to address the most significant issues.
In the area of analytical method validation and instrument performance qualification, principles and risk-based orientation, and science-based policies and standards, are the ultimate driving forces in a risk-based approach to these activities.
1. Risk-based orientation. To comply with the new guiding regulatory principle to provide the most effective public health protection, regulatory agencies and pharmaceutical companies must match their level of effort against the magnitude of risk. Resource limitations prevent uniform intensive coverage of all pharmaceutical products and production.
2. Science-based policies and standards. Significant advances in the pharmaceutical sciences and in manufacturing technologies have occurred over the last two decades. Although this knowledge has been incorporated in an ongoing manner, the fundamental nature of the changes dictates a thorough evaluation of the science base to ensure that product quality regulation not only incorporates up-to-date science but also encourages further advances in technology. Recent science can also contribute significantly to assessment of risk.
Related directly or indirectly to implementation of the risk-based approach to pharmaceutical quality, the following guidance affecting the analytical method and instrument qualification had been either initiated or implemented.
FDA 21 Code of Federal Regulations (CFR) Part 11: Electronic Records Requirements. The final guidance for industry Part 11, Electronic Records, Electronic Signatures: Scope and Application, clarifies the scope and application of the Part 11 regulation and provides for enforcement discretion in certain areas. The guidance explains the goals of this initiative, removes barriers to scientific and technological advances, and encourages the use of risk-based approaches.
ICH (International Conference on Harmonization) Q9: Risk Management. The goal of the guidance is to manage risk to patients, based on science, from information on the product, process, and facility. The level of oversight required is commensurate with the level of risk to patients and the depth of product and process understanding.
FDA Guidance for Industry PAT: A Framework for Innovative Pharmaceutical Manufacturing and Quality Assurance. This guidance is intended to encourage the voluntary development and implementation of innovative pharmaceutical manufacturing and quality assurance technologies. The scientific, risk-based framework outlined in this guidance, process analytical technology (PAT), helps pharmaceutical manufacturers design, develop, and implement new and efficient tools for use during product manufacture and quality assurance while maintaining or improving the current level of product quality assurance. It also alleviates any concerns that manufacturers may have regarding the introduction and implementation of new manufacturing technologies.
FDA Guidance for Industry: Quality Systems Approach to Pharmaceutical cGMP Regulations. One of the objectives of this guidance is to provide a framework for implementing quality by design, continual improvement, and risk management in the drug manufacturing process.
FDA Guidance for Industry INDs: cGMP for Phase 1 Investigational Drugs. This guidance recommended that sponsors and producers of phase 1 material consider carefully risks in the production environment that might adversely affect the resulting quality of an investigational drug product.
Implementation of a risk-based approach to analytical method validation and performance verification should be done simultaneously and not in isolation. It is only through a well-thought-out plan on the overall laboratory system of instrument performance verification that quality data for analytical method validation will be obtained. The laboratory will subsequently be able to support the manufacture of either clinical trial materials or pharmaceutical products for patients. Details of risk-based approaches to phase appropriate analytical method validation and performance verification are presented in subsequent chapters.
2 Regulatory Requirements for Performance Verification of Instruments
System validation requirements are specified in many different sources, including 21 CFR Part 58 [good laboratory practice (GLP)], 21 CFR Parts 210 and 211 (cGMP) [1], and more recently, in the GAMP 4 guide [2]. GLP, and GMP/cGMP are often summarized using the acronym GXP. Current GXP regulations require that analytical instruments be qualified to demonstrate suitability for the intended use. Despite the fact that instrument qualification is not a new concept and regulated firms invest a lot of effort, qualification-related deviations are frequently cited in inspectional observations and in warning letters by regulatory agencies such as the FDA and its equivalents in other countries. In common terms, the objective of qualification is to establish documented evidence that a system has been designed and installed according to specifications and operates in such a way that it fulfills its intended purpose.
GLP makes the following provisions in 21 CFR 58.63 about maintaining, calibrating, and testing equipment:
- Equipment is to be adequately inspected, cleaned, maintained, calibrated, and tested.
- Written standard operating procedures (SOPs) are required for testing, calibration, and maintenance.
- Written records are to be maintained for all inspection, maintenance, calibration, and testing.
cGMP makes the following provisions in 21 CFR 211.68(a):
- Automatic equipment, including computers, that will perform a function satisfactorily may be used.
- Equipment is to be calibrated, inspected, or checked routinely according to a written program designed to assure proper performance.
- Written records of calibration checks and inspections are to be maintained.
Many validation professionals in regulated firms are not sure what exactly to qualify or requalify, test, and document. How much testing is enough? Unlike analytical method validation, there were no clear standards for equipment qualification. The United States Pharmacopeia (USP) has addressed this issue by publishing General Chapter (1058} on analytical instrument qualification (AIQ) [3,4]. The USP establishes AIQ as the basis for data quality and defines the relationship to analytical method validation, system suitability testing, and quality control checks. Similar to analytical method validation, the intent of AIQ is to ensure the quality of an instrument before conducting any tests. In contrast, system suitability and quality control checks ensure the quality of analytical results right before or during sample analyses.
3 General Approach to Instrument Performance Qualification
Testing is one of the most important analytical measures for system developers and system users when verifying that a system fulfills the defined system requirements and is fit for the intended purpose. Generally, the fitness of systems for the intended purpose (i.e., their quality) needs to be ensured through constructive and analytical measures. Constructive measures are defined in terms of recognized professional engineering practices and include formal design methodologies that typically follow a life-cycle approach. System qualification follows a structured approach that uses test cases and test parameters based on a scientific and risk-based analysis. Defining and executing these tests typically require the use of metrology.
Other analytical measures include trending analysis of metrics such as error rates, formal methods of failure analysis, and formal reviews and inspections. Testing and the associated collection of documented evidence on the system test activities are key tasks of quality assurance. The documented evidence comprises test planning, test execution, test cases, and test results, all of which must be traceable to the requirements documented in various levels of specification documents (i.e., user requirements specification, functional specifications, design specifications, test specifications, etc.).
3.1 Definition of Terms
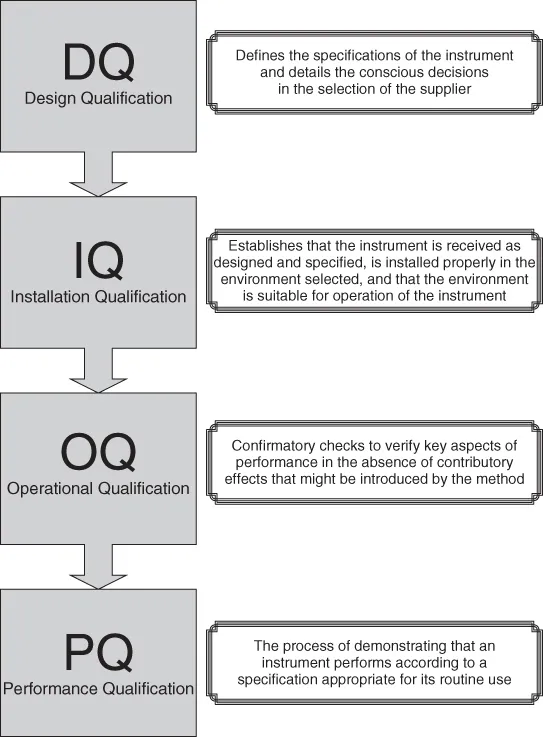
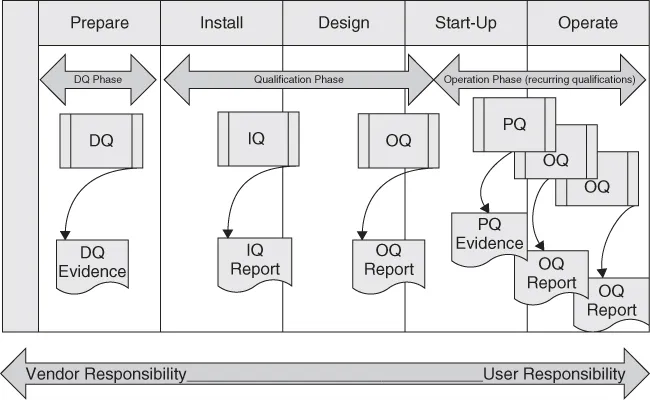
Many different definitions are used for the relevant terms in the area of equipment qualification. Not all of them are identical. For the sake of this chapter, we use the terms design qualification (DQ), installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ), in line with the definitions originally published by the Valid Analytical Measurement Instrument Working Group (see Figure 1). Similar system qualification approaches are discussed thoroughly in GAMP (Good Automated Manufacturing Practice) Forum publications and in USP General Chapter (1058). DQ, IQ, OQ, and PQ constitute important phases that result in key deliverables during the overall validation activities necessary over a system's life cycle (see Figure 2).
Design Qualification
During DQ, the functional and operational specifications of an instrument need to be defined and documented. DQ is an important decision-making tool for selecting the best system and supplier. The right type of equipment is selected for specific tasks, and the supplier's ability to meet and reproduce these performance criteria consistently through appropriate quality processes in design, development, manufacturing, and support is crucial for efficacy and risk mitigation. DQ is primarily the user's responsibility, because this is the only logical place to define site requirements. The supplier, however, typically needs to provide materials such as technical specifications and other documents relevant to system validation. This includes evidence on processes that are critical to quality, including the life-cycle methodology. DQ focuses on specifications, design documentation, requirements traceability from design to test, corrective action procedures, impact analyses, test plans, and test evidence. DQ responds to a requirement originally defined in GLP (21 CFR Part 58.61) that mandates that appropriate...