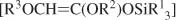![]()
CHAPTER 1
Reactions of Aldehydes and Ketones and their Derivatives
A. C. KNIPE
Faculty of Life and Health Sciences, University of Ulster, Coleraine
Formation and Reactions of Acetals and Related Species
Reactions of Glucosides and Nucleosides
Formation and Reactions of Nitrogen Derivatives
Imines: Synthesis, Tautomerism, Catalysis
The Mannich and Nitro-Mannich Reactions
Addition of Organometallics
Oxidation and Reduction of Imines
Iminium Species
Imine Cycloadditions
Other Reactions of Imines
Oximes, Hydrazones, and Related Species
C–C Bond Formation and Fission: Aldol and Related Reactions
Regio-, Enantio-, and Diastereo-selective Aldol Reactions
Intramolecular Aldols
Mukaiyama and Vinylogous Aldols
Nitrile/Nitro/Nitroso Aldols
Other Aldol-type Reactions
Pinacol- and Benzoin-type Coupling
The Baylis-Hillman and its Aza and Morita Variants
Allylation and Related Reactions
Alkynations
Michael Additions
Other Addition Reactions
General and Theoretical
Addition of Organozincs
Addition of Other Organometallics
Grignard-type Reactions
Hydrocyanation and Cyanosilylation
Hydrosilylation and Hydrophosphonylation
Miscellaneous Additions
Enolization and Related Reactions
Oxidation and Reduction of Carbonyl Compounds
Regio-, Enantio-, and Diastereo-selective Reduction Reactions
Oxidation Reactions
Other Reactions
References
Formation and Reactions of Acetals and Related Species
A water-soluble self-assembled host molecule [Ga4L6]12− has been found to catalyse the hydrolysis of orthoformates HC(OR)3 in basic solution with marked rate accelerations of up to 3900 (R = Pr) relative to the uncatalysed reaction.1 Enzyme-like Michaelis-Menten kinetics apply and 13C labeling experiments have helped to establish that the neutral substrate is encapsulated in the host in the resting state; this is consistent with the more negative entropy of activation found for the catalytic process. The solvent isotope effects k;(H2O)/k(D2O) = 1.6 is indicative of an A-SE2 mechanism with rate-limiting proton transfer, in contrast with the A1 mechanism, for the catalysed reaction which involves rate-limiting decomposition of the protonated substrate.
Pyridinium salt derivatives have been found to catalyse (at 0.1% loading) acetalization reactions of both aldehydes and ketones in methanol at room temperature more efficiently than Brønsted acids of pKa = 22.2
A study of the effects of the strength of the nucleophile (Nu-SiMe3) on the stereochemical outcome of substitution reactions of cyclic acetals (in CH2Cl2 in the presence of Me3SiOTf or BF3OEt2) has revealed a continuum of mechanisms. Stereoselective SN1 mechanisms (via the oxocarbenium ion intermediate) occur with weak and moderate nucleophiles and poor leaving groups, whereas strong nucleophiles adopt unselective diffusion-limited SNl and SN2 pathways.3
A highly enantioselective (up to 99% ee) and diastereoselective aldol-type reaction of β-keto esters with acetals has been achieved under the catalytic influence of chiral cationic Pd(II)-and Pt(II)-binap complexes which can act as acid/base catalysts, with simultaneous activation of both the nucleophile (as chiral metal enolate) and acetal;4the enantioselectivity is apparently dependent on conversion of the acetal into the oxonium ion, by protonation under the acidic conditions used.
A Lewis base-catalysed diastereoselective and enantioselective glycolate aldol reaction has been developed whereby a range of aldehydes can be converted to both
syn-and
anti-1,2-diols under the same catalytic system by adjusting the size of the silyl ketene acetal
used as the nucleophilic component; these Mukaiyama-type aldol reactions are conducted In CH
2Cl
2 at −78°C in the presence of SiCl
4,
i-Pr
2NEt and a bisphosphoramide catalyst.
5The Brønsted acid catalysed aza-Ferrier reaction of N-Boc-2-(l,3-dienyloxy) pyrrolidines has been found to proceed via heterolytic C–O cleavage to give the iminium alkoxlde, from which the corresponding α-(N-Boc-2-pyrrolidinyl)aldehyde is formed in excellent yield and high α-regioselectivity by C–C bond formation.6
Reactions of Glucosides and Nucleosides
Nucleophilic substitution reactions of 2-phenylthio-substituted carbohydrate acetals and related systems have been reviewed, with particular reference to episulfonium ions vs oxocarbenium ions as reactive intermediates in stereocontrolled glycosylation reactions.7
Mechanistic study (using kinetic experiments, Hammet plots, DFT calculations and 11B NMR) of regioselective borane reductive openings of cyclic acetals has established that the outcome depends on the electrophile with which the more electron-rich oxygen associates, and this can be altered by Lewis acid activation of the borane.8
A plausible mechanism has been suggested to account for α-selective glucosylation which allowed the synthesis of the tetrasaccharide [Glcα1 → 2Glcα1 → 3Glcα1 → 3Man], enabled by the synergistic effect of combined etheral and halogenic solvents.9
Hydrolysis of toxic 7-hydroxycoumarin glucosides and other aryl and alkyl glucosides catalysed by modified α- and β-cyclodextrin dicyanohydrins has been found to follow Michaelis-Menten kinetics and to display rate increases of up to kcat/kuncat = 7569 (for the hydroxycoumarin glucoside).10
Results of a DFT study of suitable analogues have explained the relative O(3)/O(4) reactivities previously reported for reaction of glycosyl donors with both α- and β-methyl glycosides of N-dimethyrmaleoyl (DMM) glucosamine acceptors protected at O(6).11 The preferential or exclusive substitution at O(3) for the α-anomers and at O(4) for the β-anomers has been attributed to the different alignments for the DMM ring: for the β-anomers the ring is parallel to the C(2)–H(2) bond for steric reasons, whereas for the α-anomers it is tilted such that a strong hydrogen bond between one of its carbonyl groups and HO(3) makes O(3) more reactive.
A study of potential neighbouring group participation by non-vicinal esters in glycosylation reactions has found that glycopyranosylation Is aided by intermediate formation of a 1,3-O-cyclic carbonate ester, with loss of a t-butyl cation from a t-butoxycarbonyl ester group axially substituted at C(3), but that the corresponding 3-O-equatorial, 4-O-axial and -equatorial, and 6-O carbonates do not undergo intramolecular reaction under typical glycosylation conditions.12 Galactopyranosylation reactions of a 4-O-(2-carboxy)benzoate ester and a 4-O-(4-methoxybenzoate) ester also failed to exhibit neighbouring group participation; this was particularly clear for the latter, for which 18O quenching failed to detect bridging intermediates. The unesterified hydroxyl groups were protected as benzyl ethers.
Formation and Reactions of Nitrogen Derivatives
Imines: Synthesis, Tautomerism, Catalysis
Isomerization of the imine derived from benzylamine an...