
Statistical and Machine Learning Approaches for Network Analysis
- English
- ePUB (mobile friendly)
- Available on iOS & Android
Statistical and Machine Learning Approaches for Network Analysis
About this book
Explore the multidisciplinary nature of complex networks through machine learning techniques
Statistical and Machine Learning Approaches for Network Analysis provides an accessible framework for structurally analyzing graphs by bringing together known and novel approaches on graph classes and graph measures for classification. By providing different approaches based on experimental data, the book uniquely sets itself apart from the current literature by exploring the application of machine learning techniques to various types of complex networks.
Comprised of chapters written by internationally renowned researchers in the field of interdisciplinary network theory, the book presents current and classical methods to analyze networks statistically. Methods from machine learning, data mining, and information theory are strongly emphasized throughout. Real data sets are used to showcase the discussed methods and topics, which include:
- A survey of computational approaches to reconstruct and partition biological networks
- An introduction to complex networks—measures, statistical properties, and models
- Modeling for evolving biological networks
- The structure of an evolving random bipartite graph
- Density-based enumeration in structured data
- Hyponym extraction employing a weighted graph kernel
Statistical and Machine Learning Approaches for Network Analysis is an excellent supplemental text for graduate-level, cross-disciplinary courses in applied discrete mathematics, bioinformatics, pattern recognition, and computer science. The book is also a valuable reference for researchers and practitioners in the fields of applied discrete mathematics, machine learning, data mining, and biostatistics.
Tools to learn more effectively

Saving Books

Keyword Search

Annotating Text

Listen to it instead
Information
1.1 Introduction
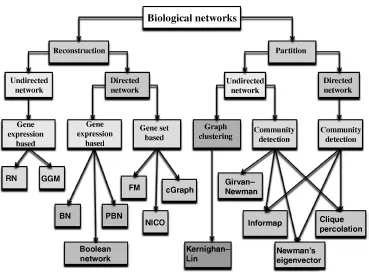
Table of contents
- Cover
- Title Page
- Copyright
- Dedication
- Preface
- Contributors
- Chapter 1: A Survey of Computational Approaches to Reconstruct and Partition Biological Networks
- Chapter 2: Introduction to Complex Networks: Measures, Statistical Properties, and Models
- Chapter 3: Modeling for Evolving Biological Networks
- Chapter 4: Modularity Configurations in Biological Networks with Embedded Dynamics
- Chapter 5: Influence of Statistical Estimators on the Large-Scale Causal Inference of Regulatory Networks
- Chapter 6: Weighted Spectral Distribution: A Metric for Structural Analysis of Networks
- Chapter 7: The Structure of an Evolving Random Bipartite Graph
- Chapter 8: Graph Kernels
- Chapter 9: Network-Based Information Synergy Analysis for Alzheimer Disease
- Chapter 10: Density-Based Set Enumeration in Structured Data
- Chapter 11: Hyponym Extraction Employing a Weighted Graph Kernel
- Index
Frequently asked questions
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app