![]()
SECTION IV
Nutrition, Hypertension, and Cardiovascular Disease
![]()
CHAPTER 33
From Abdominal Adiposity to Hypertension: Mechanisms and Clinical Implications
Albert P. Rocchini
University of Michigan, Ann Arbor, MI, USA
Introduction
Obesity, and visceral adiposity in particular, is often associated with systemic hypertension. This chapter describes the evidence that links obesity and visceral adiposity in particular, to hypertension, the potential mechanisms relating visceral obesity and the development of hypertension, and the clinical implications of the association of visceral obesity and hypertension.
Relationship between Obesity and High Blood Pressure
Epidemiologic Studies Linking Obesity to Hypertension
The association between obesity and hypertension has been recognized since the early 1900s. Many large epidemiologic studies document the association between increasing body weight and an increase in blood pressure [1–3]. For example, the Framingham study [1] documented that the prevalence of hypertension in obese individuals was twice that of average-weight individuals. This relationship was seen in all age-groups of both women and men. Based on this and many other population based studies [1–3], we know that there is a very strong association between obesity and hypertension in both sexes, in all age-groups, and for virtually every geographical and ethnic group.
Relationship of Weight Gain to Blood Pressure Level
There have been no studies in humans that have looked at the effect of weight gain on blood pressure. However, in the dog, it has been shown that weight gain is directly associated with an increase in blood pressure. In 1938, Cash and Wood demonstrated that weight gain caused dogs with renal vascular hypertension to further increase their blood pressure. More recently, Rocchini et al. [4] and Hall et al. [5] reported that normal mongrel dogs fed a high-fat diet gained weight and developed hypertension. In these dogs, the hypertension was associated with sodium retention, hyperinsulinemia, and activation of the sympathetic nervous system.
Effect of Weight Loss on Blood Pressure Level
Weight loss is associated with a lowering of blood pressure. Many clinical trials that have been published since the late 1970s have clearly documented the blood pressure-lowering effect of weight loss [6–8]. For example, the Hypertension Prevention Trial [7] documented that in individuals with borderline elevations in blood pressure a mean weight loss of 5 kg was associated with as much as a 5/3 mm Hg decrease in blood pressure. Thus, based on numerous weight loss studies, calorie restriction and weight loss are associated with a reduction in blood pressure. In addition, it is clear that even modest weight loss (i.e., 10% of body weight) improves blood pressure, and many individuals achieve normal blood pressure levels without attaining their calculated ideal weight.
A limitation with the use of studies documenting that weight loss is associated with a reduction in blood pressure is that most studies do not address the long-term effect of weight change on blood pressure in subjects who are again placed on unrestricted diets. Dornfield et al. [9] reported that over a follow-up of 1–4 years after weight loss, changes in blood pressure still correlate with changes in body weight. However, bariatric surgery data suggests that long-term weight loss may not reduce the incidence of hypertension. Sjostrom et al. [10] compared the incidence of hypertension and diabetes in 346 patients undergoing gastric surgery with 346 obese control subjects who were matched on 18 variables. After 8 years, the surgical group had maintained a 16% weight loss, whereas the control subjects had a 1% weight gain. These investigators demonstrated that the weight reduction in the surgical group had a dramatic effect on the 8-year incidence of diabetes but had no effect on the 8-year incidence of hypertension. They previously documented that surgical weight loss positively affected blood pressure at 2 and 4 years of follow-up, but that this effect on blood pressure was lost after 8 years of follow-up despite a maintained 16% weight reduction. These authors have speculated “that remaining obesity in the surgically treated patients could have induced a reappearance of hypertension during the course of the study independent of initial body weight and initial weight loss.” Therefore, Sjostrom’s study suggests that a relapse of hypertension after surgically induced weight loss does occur despite the maintenance of significant long-term weight loss and that the pathogenesis of recurrent hypertension is not well understood [10].
Effect of Body Fat Distribution on Blood Pressure
The definition of obesity also contributes to the controversy regarding the independence of obesity as an etiological determinant of hypertension. Obesity is defined not just as an increase in body weight but rather as an increase in adipose tissue mass. Adipose tissue mass can be estimated by multiple techniques such as skinfold thickness, body mass index (BMI, [weight in kg]/[height in meters]2), hydrostatic weighing, bioelectrical impedance, water dilution methods, computed tomography (CT), and magnetic resonance imaging (MRI). In most clinical studies, BMI is usually used as the index of adiposity. Obesity is generally defined as a BMI of greater than 30kg/m2. In 1956, Jean Vague reported that the cardiovascular and metabolic consequences of obesity were greatest in individuals whose fat distribution pattern favored the upper body segments. Since that observation, several population based studies have demonstrated that abdominal obesity is a more important cardiovascular risk factor than BMI alone [3,11], thus, suggesting that increased visceral adipose tissue (VAT) as opposed to subcutaneous adipose tissue (SAT) relates better to the development of systemic hypertension. For example, Fox et al. [3] demonstrated in 3,001 participants from the Framingham Heart Study, that although both SAT and VAT are associated with the prevalence of hypertension, only VAT provides significant information above and beyond percent fat and waist circumference. Many investigators have demonstrated that the association of obesity to increased cardiovascular risk is also primarily related to abdominal adiposity [11]. Finally, in dogs that develop hypertension by being fed a high-fat diet, the increase in their abdominal circumference is significantly greater than that of their thoracic circumference [12]. MRI studies in these fat-fed dogs demonstrate a marked increase in omental and subcutaneous fat [13]. We also have preliminary data in dogs fed a high-fat diet that demonstrates a stronger relationship between the increase in blood pressure and the increase in abdominal circumference as compared to the increase in body weight.
Mechanism Whereby Obesity Might Cause Hypertension
Although there is a strong relation between hypertension, obesity, and abdominal obesity in particular, the mechanism whereby increased adiposity leads to the development of hypertension has not been completely elucidated. It is clear that obesity hypertension directly relates to abnormal renal sodium handling and that this alteration in sodium handling is predominately mediated through activation of the sympathetic nervous system and to a lesser extent through activation of the renin-angiotensin-aldosterone system. However, what is less clear is how obesity initiates the activation of the sympathetic nervous system.
Abnormal Renal Sodium Handling and Obesity Hypertension
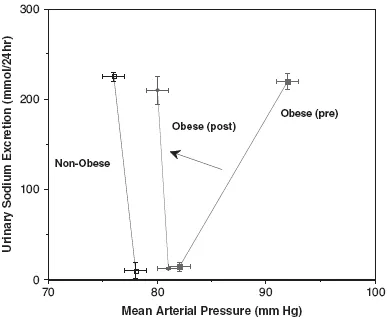
There is ample human and animal data linking obesity hypertension to fluid retention. Many investigators have reported that obesity is associated with an increased cardiac output and blood volume. Rocchini et al. [8] demonstrated that prior to weight loss, the blood pressure of a group of obese adolescents was very sensitive to dietary sodium intake; however, after weight loss, the obese adolescent lost their blood pressure sensitivity to sodium. These investigators demonstrated that when compared to nonobese adolescents, the obese adolescents have a renal-function relation (plot of urinary sodium excretion as a function of arterial pressure) that has a shallower slope. The renal-function relationship is also normalized by weight loss (Figure 33.1).
In addition, abdominal adiposity is associated with altered renal tubular sodium handling. Strazzullo et al. [14,15] measured proximal and distal fractional sodium reabsorption in 702 participants of the Olivetti Heart Study. These investigators demonstrated in adult men that the metabolic syndrome was associated with an increased rate of proximal tubular sodium reabsorption. Similarly Barbato et al. [16] demonstrated that increased proximal sodium reabsorption is associated with the metabolic syndrome in both white men and women; however, this relationship is not seen in people of African or South Asian origin, even though these two ethnic groups have a greater degree of insulin resistance and central adiposity. These investigators could not prove why alter proximal tubular sodium reabsorption was not observed in individuals of African or Asian origin; however they speculated that it could be due to differences in habitual sodium intake, differences in genetic background.
There is also animal data that suggests that sodium retention is associated with obesity hypertension. In a dog model of obesity-induced hypertension, Rocchini et al. [4] demonstrated that during the first week of the high-fat diet, the increase in sodium retention appeared to best relate to an increase in plasma norepinephrine (NE) activity, whereas during the latter weeks of the high-fat diet, an increase in plasma insulin appeared to be the best predictor of sodium retention. Rocchini also demonstrated that the hypertension associated with weight gain in the dog occurs only if adequate salt is present in the diet. Hall et al. [17] demonstrated that obesity-induced hypertension in the dog is associated with increased renal tubular sodium reabsorption since marked sodium retention occurred despite large increases in glomerular filtration and renal plasma flow. Ganger et al. [18] demonstrated that dogs fed a high-fat diet develop an abnormal renal pressure-natriuresis relationship similar to that observed in obese adolescents.
The relationship between urinary sodium excretion and mean arterial pressure can be altered by intrinsic and extrinsic factors that are known to affect the ability of the kidney to excrete sodium. Factors that produce alterations in the renal-function curves are constriction of the renal arteries and arterioles, changes in glomerular filtration coefficients, changes in the rate of tubular reabsorption, reduced renal mass, and changing levels of renin-angiotensin activation, aldosterone, vasopressin, insulin, sympathetic nervous system activation, and atrial natriuretic hormone. Although both obese humans and animals can have compression of the kidney by the surrounding fat and that fat may penetrate the renal hilum into the sinuses surrounding the renal medulla [19], it is unlikely that fat-based structural changes in the kidney is the major pathophysiological cause of the renal sodium retention associated with obesity. Based on both human and animal data, activation of the renin-angiotensinaldosterone systemand that of the sympathetic nervous system are the most likely factors responsible for the altered renal function curves observed in obesity.
Renin-Angiotensin-Aldosterone System
The renin-angiotensin-aldosterone system is an important determinant of efferent glomerular arteriolar tone, and tubular sodium reabsorption. Its activity is modulated by dietary salt ingestion, blood pressure, and the sympathetic nervous system. Therefore, alterations in the renin-angiotensin-aldosterone system could be expected to alter pressure natriuresis. Enhanced activity of the renin-angiotensin-aldosterone system has been reported in obese humans and dogs [18,20,21]. Granger et al. [18] reported that plasma renin activity is 170% higher in obese dogs than in control dogs.
Aldosterone concentrations have been demonstrated to be abnormal in both human and animal obesity [4,21]. For example, Rocchini et al. [21] measured supine and 2-hour upright plasma renin activity and aldosterone in 10 nonobese and 30 obese adolescents before and after a 20-week weight loss program. The obese adolescents had significantly higher supine and 2-hour upright aldosterone concentrations. Although plasma renin activity was not significantly different between the two groups of adolescents, they observed that a given increment in plasma renin activity produced a greater increment in aldosterone in the obese adolescents. Compared with an obese control group, weight loss resulted in both a significant decrease in plasma aldosterone and a significant decrease in the slope of the posture-induced relation between plasma renin activity and aldosterone. Goodfriend and Calhoun [22] suggested that increased plasma free fatty acids (FFAs) produced in obese individuals may stimulate aldosterone production independent of renin.
Insulin also has been shown to influence the renin-angiotensin-aldosterone system in both normal subjects and in patients with diabetes. For example, Rocchini et al. [20] measured the increase in plasma aldosterone after graded increases in intravenous angiotensin II before and after euglycemic hyperinsulinemia in seven chronically instrumented dogs. Euglycemic hyperinsulinemia resulted in a significantly greater (p < .01) change in the angiotensin II–stimulated increments of plasma aldosterone than was observed when angiotensin II was administered alone. However, there was no dose-dependence of insulin’s effect on angiotensin II–stimulated aldosterone. In addition, although weight gain significantly increased angiotensin II–stimulated aldosterone, with hyperinsulinemia the response was not significantly different than that observed in the dogs prior to weight gain.
Despite these results suggesting that obesity is associated with significant alterations in the renin-angiotensin-aldosterone system, Hall et al. [17] demonstrated that weight-related changes in blood pressure can occur in dogs independent of changes in angiotensin II, and de Paula et al. [23] demonstrated that the aldosterone antagonist, eplerenone, attenuated but did not prevent the sodium retention and hypertension associated with feeding dogs a high fat diet. Thus, although the renin-angiotensinaldosterone system plays an important role in the pathogenesis of obesity hypertension, it is neither the major nor the sole mechanism responsible for the altered renal pressure–natriuresis relationship observed in obesity.
Sympathetic Nervous System
For over 20 years, it has been recognized that diet affects the sympathetic nervous system. Fasting suppresses sympathetic nervous system activity, whereas overfeeding with either a highcarbohydrate or a high-fat diet stimulates the sympathetic nervous system [24,25]. It is believed that the physiological consequence of the link between dietary intake and sympathetic nervous system activity is to regulate energy expenditure in a hope to maintain weight homeostasis. Landsberg [25] suggested that in obese individuals the sympathetic nervous system is chronically activated in an attempt to prevent further weight gain and that hypertension is a byproduct of the overactive sympathetic nervous system. Landsberg [25] proposed that obesity produces a compensatory sympathetic activation, which contributes to the cardiovascular morbidity associated with it. Microneurography, which directly measures sympathetic traffic to skeletal muscle, has consistently shown to be increased in obesity [26]. Previous studies in obese subjects have reported a positive association between sympathetic activity and increased blood pressure. We have preliminary data demonstrating that over six weeks of feeding dogs a high fat diet that although serial plasma NE concentrations only trended toward increasing (p=.09); however, using serial NE kinetic studies, we observed a significant (p < .001) increase in the rate of NE release from the sympathetic nerve terminals (NE2). The most likely reason that we did not demonstrate a significant increase in plasma NE levels was because in addition to the increased rate of NE release from the sympathetic nerve terminals, we also observed a significant increase in NE clearance. In addition, after 6 weeks of the high-fat diet, there was a strong relationship between the increase in NE2 and the increase in arterial pressure. In the fat-fed dog, Kassab et al. [27] demonstrated that renal denervation prevents both the sodium retention and the hypertension associated with weight gain but does not prevent insulin resistance. In addition, Eikelis et al. [28], using regional analysis of NE kinetics, demonstrated increased renal NE spillover in obese subjects. In both animal and human studies, pharmacologic blockade of the sympathetic nervous system prevents the increase in blood pressure and sodium retention associated with obesity [29,30]. Finally, Lohmeier et al. [31] demonstrated that fat feeding of dogs causes a marked increase in activity of the protein product of the immediate early gene c-fos in the baroreceptor sympathoexcitatory cells in the rostral ventrolateral medulla, a site known to be affected by both angiotensin II and leptin. Increased gene c-fos expression in the rostral ventrolateral medulla of obese dogs supports the observations that sympathetic activity to the kidney and other vascular beds is increased in obesity hypertension [27,28]. Thus, activation of the sympathetic nervous systems appears to be one of the major factors responsible for both the altered renal-function relationship and hypertension observed in obesity. However, what is still unknown is what is the factor or factors responsible for activation of the sympathetic nervous system in obesity.
Possible Mechanisms Responsible for Activation of the Sympathetic Nervous System in Obesity
Since increased VAT appears to be the best predictor of hypertension [3], it is likely that increased sympathetic activation is related to the metabolically active adipose tissue found in the visceral region. VAT is known to secrete FFAs, adipocytokines, and inflammatory cytokines into the portal circulation. Three possible mechanisms that may be responsible for the increase in sympathetic nervous system activity associated with obesity are increased FFA levels in the portal circulation, increased adipocytokines and inflammatory cytokines, and/or increased secretion of leptin.
Increased Portal FFA and Sympathetic Activation
Increased portal FFA may increase sympathetic activity through the development of insulin resistance and hyperinsulinemia. Arner et al. [32] first suggested that the release into the portal vein of FFAs originating from visceral fat might be responsible for the development of insulin resistance. There are a number of repor...