
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
About this book
Provides hands-on knowledge enabling students of and researchers in chemistry, biology, and engineering to perform molecular simulations
This book introduces the fundamentals of molecular simulations for a broad, practice-oriented audience and presents a thorough overview of the underlying concepts. It covers classical mechanics for many-molecule systems as well as force-field models in classical molecular dynamics; introduces probability concepts and statistical mechanics; and analyzes numerous simulation methods, techniques, and applications.
Molecular Simulations: Fundamentals and Practice starts by covering Newton's equations, which form the basis of classical mechanics, then continues on to force-field methods for modelling potential energy surfaces. It gives an account of probability concepts before subsequently introducing readers to statistical and quantum mechanics. In addition to Monte-Carlo methods, which are based on random sampling, the core of the book covers molecular dynamics simulations in detail and shows how to derive critical physical parameters. It finishes by presenting advanced techniques, and gives invaluable advice on how to set up simulations for a diverse range of applications.
-Addresses the current need of students of and researchers in chemistry, biology, and engineering to understand and perform their own molecular simulations
-Covers the nitty-gritty ? from Newton's equations and classical mechanics over force-field methods, potential energy surfaces, and probability concepts to statistical and quantum mechanics
-Introduces physical, chemical, and mathematical background knowledge in direct relation with simulation practice
-Highlights deterministic approaches and random sampling (eg: molecular dynamics versus Monte-Carlo methods)
-Contains advanced techniques and practical advice for setting up different simulations to prepare readers entering this exciting field
Molecular Simulations: Fundamentals and Practice is an excellent book benefitting chemist, biologists, engineers as well as materials scientists and those involved in biotechnology.
This book introduces the fundamentals of molecular simulations for a broad, practice-oriented audience and presents a thorough overview of the underlying concepts. It covers classical mechanics for many-molecule systems as well as force-field models in classical molecular dynamics; introduces probability concepts and statistical mechanics; and analyzes numerous simulation methods, techniques, and applications.
Molecular Simulations: Fundamentals and Practice starts by covering Newton's equations, which form the basis of classical mechanics, then continues on to force-field methods for modelling potential energy surfaces. It gives an account of probability concepts before subsequently introducing readers to statistical and quantum mechanics. In addition to Monte-Carlo methods, which are based on random sampling, the core of the book covers molecular dynamics simulations in detail and shows how to derive critical physical parameters. It finishes by presenting advanced techniques, and gives invaluable advice on how to set up simulations for a diverse range of applications.
-Addresses the current need of students of and researchers in chemistry, biology, and engineering to understand and perform their own molecular simulations
-Covers the nitty-gritty ? from Newton's equations and classical mechanics over force-field methods, potential energy surfaces, and probability concepts to statistical and quantum mechanics
-Introduces physical, chemical, and mathematical background knowledge in direct relation with simulation practice
-Highlights deterministic approaches and random sampling (eg: molecular dynamics versus Monte-Carlo methods)
-Contains advanced techniques and practical advice for setting up different simulations to prepare readers entering this exciting field
Molecular Simulations: Fundamentals and Practice is an excellent book benefitting chemist, biologists, engineers as well as materials scientists and those involved in biotechnology.
Tools to learn more effectively

Saving Books

Keyword Search

Annotating Text

Listen to it instead
Information
1
Introduction – Studying Systems from Two Viewpoints
When analyzing physical, chemical, and biological systems, macroscopic and microscopic viewpoints give two seemingly different descriptions. For example, as shown in Figure 1.1, the state of a gas inside a pressurized capsule is described from a macroscopic viewpoint by a limited number of variables, including pressure, P, volume, V, and temperature, T. Depending on the nature of the gas and the macroscopic state, these variables are related by an equation of state such as the ideal gas law, PV = nRT. This description of the gas includes the mechanical variables pressure and volume, and a nonmechanical variable, temperature. In the macroscopic view, the behavior of the gas is governed by the laws of thermodynamics and no reference is made to the molecular structure of the gas. Indeed, thermodynamics was developed in the mid‐nineteenth century before atomic theory of matter was widely accepted by physicists. The thermodynamic description is used for macroscopic samples with micrometer or larger length scales over long times.
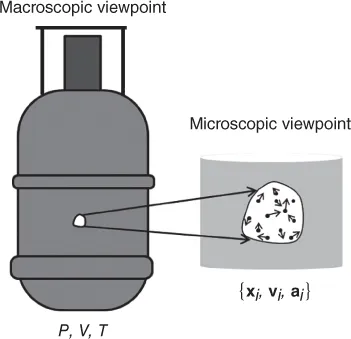
Figure 1.1 Macroscopic and microscopic viewpoints of a gas system involve different variables, length, and time scales.
From a microscopic (atomic) viewpoint, a gas is a collection of a large number (in the order of 1023) of molecules, moving randomly at high speeds, each with a specified position, velocity, and acceleration at any given time. The microscopic description of the gas uses only mechanical variables that obey the laws of classical mechanics. The details of atomic/molecular structures and interactions, along with the application of Newton's equation of motion, determine how positions and velocities of the molecules of the gas change with time. The laws of conservation of energy, linear momentum, and angular momentum constrain the mechanical variables throughout the process. Knowledge of mechanical variables at any time allows the calculation of these variables at all times in the future and past (neglecting considerations of classical nonlinear systems and quantum mechanics) and classical mechanics is therefore deterministic with regard to mechanical variables. The classical mechanical microscopic description does not include macroscopic variables such as temperature and entropy, which are used to describe macroscopic systems.The microscopic description is used to describe phenomena on length scales of the order of nanometers and time scales of the order of nanoseconds.
How are these dual descriptions of physical systems reconciled, and why is there such a discrepancy in the length and time scales between these two viewpoints? How do nonmechanical variables get introduced into analysis of system properties in the macroscopic viewpoint, if these macroscopic variables do not appear in the underlying microscopic description of the system, which is supposedly more fundamental? The answers to these questions form the context of molecular simulation.
In his book “What is Life?” Erwin Schrödinger asks the question every student has wondered when first introduced to atoms: Why are atoms so small? [269] Our daily experience captures length scales as small as millimeters, while atoms and molecules with dimensions in the nanometer range are smaller by factors of 10−7/10−8 than any phenomena we experience directly. Even the smallest bacteria have dimensions in the micrometer range, which gives them a length range larger by a factor of 104 compared to atoms and molecules. Why are there such discrepancies in length and time scales between atoms and the macroscopic phenomenon of life?
Schrödinger argues that since atoms are fundamental building blocks of matter, this is not the correct question to ask. The question should be reframed as “Why are we, as living organisms, so much larger than atoms?” or “Why are there so many atoms and molecules in cells and more complex organisms?” Stated differently, the question can be “Why is Avogadro's number so large?” The answers to these questions determine how new system properties emerge as we transition from the microscopic mechanical descriptions of systems to the macroscopic thermodynamic description of large systems.
The connection between microscopic and macroscopic descriptions is made by invoking probability theory arguments in statistical mechanics. Relatively simple microscopic systems such as ideal gases are amenable to analytical statistical mechanical analysis, and explicit formulas relating microscopic mechanical properties of the gas molecules to macroscopic thermodynamic variables can be derived. For more complex microscopic systems, molecular simulations (using numerical computations) within the framework of molecular dynamics or Monte Carlo simulations are performed and statistical mechanical relations relate the averages of molecular properties to macroscopic observables of these systems.
This book gives an introduction to the microscopic molecular dynamics and Monte Carlo simulation methods for calculating the macroscopic properties of systems. Even in cases where the goal is a purely microscopic mechanical study of the system, there are usually macroscopic constraints imposed on the system by the environment. For example, the conditions of constant physiological temperature and ambient pressure imposes constraints on molecular simulations when studying the interaction of a drug candidate with an enzyme binding site in aqueous solution. These constraints impose nonmechanical conditions on the microscopic description of the system that must be applied correctly when simulating molecular behavior.
Chapter 2 gives a brief overview of classical mechanics used to describe the motion of atoms and molecules in microscopic systems. We start from simple physical systems for which analytical solutions of the classical Newtonian equations are available and move to complex multiatom systems for which numerical methods of solution (namely, finite difference methods) are needed. The concept of phase space trajectory, which describes the dynamics of these systems, is introduced.
Solving Newton's laws of motion for a molecular system requires knowledge of the forces acting between atoms. In Chapter 3, the quantum mechanical basis for determining the interatomic forces within and between molecules and their classical approximations are described. A description of classical force fields used in molecular simulations of chemical and biological systems follows.
Having introduced numerical methods to solve the classical equations of motion and the microscopic forces acting between atoms, the next step is the introduction of specialized techniques needed to make molecular simulations feasible. These techniques, which include the use of periodic boundary conditions, potential cutoffs for short range forces, and Ewald summation methods for long range electrostatic forces, are discussed in Chapter 4.
In Chapters 5 and 6, we introduce concepts from probability theory that describe how to predict and analyze behaviors of complex systems on which we have too little or too much information. Concepts of probability theory as applied to mechanical systems form the framework for statistical mechanics. Relations of probability theory and statistical mechanics must be considered to correctly run a molecular simulation and to ensure that the molecular level system is treated in a manner consistent with macroscopic conditions imposed on the system. Molecular simulation results can then be subjected to further statistical mechanical analysis or be used to get direct microscopic insight into phenomena of interest. In Chapter 5, the principles of probability theory are applied to non-interacting systems, while in Chapter 6, the concept of the ensemble of systems is introduced, which allows probabilistic analysis of systems that include intermolecular interactions. The classical expressions for the probability distributions for different ensembles are the constraints that molecular simulations must satisfy.
Chapters 7 and 10 cover specialized molecular simulation techniques for imposing specific values of macroscopic thermodynamic variables in a simulated system. In Chapter 7 methods of correctly imposing constant pressure (Andersen barostat) and constant temperature (Nosé–Hoover thermostat) on systems of molecules in molecular simulations are described. In Chapter 10, the grand canonical Monte Carlo simulation method for imposing the condition of fixed chemical potential and temperature is described.
Chapter 8 and 9 treat the extraction and analysis of structural/thermodynamic properties and dynamic properties using molecular dynamics simulations, respectively. Selected examples from a large body of simulation work are outlined.
Throughout the book, we will emphasize an appreciation of time, length, and energy scales of molecular processes including molecular translations, vibrational processes, and bulk fluid motions.
Many excellent books, articles, and websites on mechanics, probability theory, statistical mechanics, and molecular simulation methods are available and have been cited in the references. These have undoubtedly influenced the presentation of the material here and explicit citations are given in different sections as appropriate. A large body of work on molecular dynamics and Monte Carlo methods is available and only a small sample of topics could be covered here. Important and groundbreaking work by many experts has not been discussed, and this is a reflection of the limited scope of this book rather than the importance of the work. Contributions of researchers from the past and present are gratefully acknowledged, although they are not mentioned individually here.
A further point is that many important advanced modern topics are not covered in this book as they are beyond the scope of this introductory discourse. For example, free energy methods, biased Monte Carlo sampling, and methods of high‐performance computing used in molecular simulations are not discussed. It is hoped that the introductory material in this book provides a launching pad for the study of these advanced topics. For more advanced users, it is hoped that this book can provide a useful overview and some intuitive understanding of methods that go into molecular simulations.
Table of contents
- Cover
- Table of Contents
- Preface
- 1 Introduction – Studying Systems from Two Viewpoints
- 2 Classical Mechanics and Numerical Methods
- 3 Intra‐ and Intermolecular Potentials in Simulations
- 4 The Mechanics of Molecular Dynamics
- 5 Probability Theory and Molecular Simulations
- 6 Statistical Mechanics in Molecular Simulations
- 7 Thermostats and Barostats
- 8 Simulations of Structural and Thermodynamic Properties
- 9 Simulations of Dynamic Properties
- 10 Monte Carlo Simulations
- Index
- End User License Agreement
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Molecular Simulations by Saman Alavi in PDF and/or ePUB format, as well as other popular books in Technology & Engineering & Materials Science. We have over one million books available in our catalogue for you to explore.