![]()
CHAPTER 1
Introduction to ChIP-seq
Borbala Mifsud
CHROMATIN immunoprecipitation followed by high-throughput sequencing (ChIP-seq) is amongst the most widely used methods in molecular biology. It aims to determine the genomic sites that interact with a protein of interest. ChIP-seq can be used to identify transcription factor (TF) binding sites or broader patterns of post-translational histone modifications, referred to as histone marks, underlying regulatory elements. This method is therefore essential for deepening our understanding of transcriptional regulation.
1.1 CHIP-SEQ EXPERIMENT
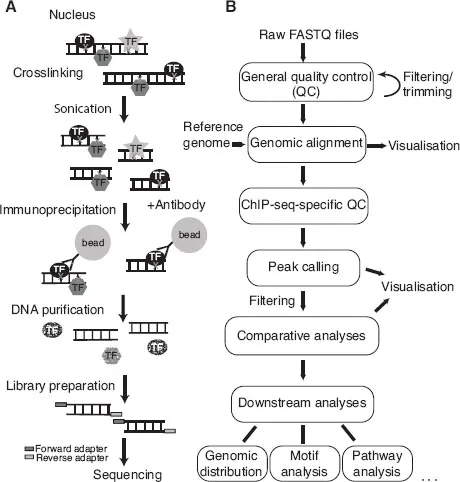
ChIP-seq is an experiment based on antibody immunoprecipitation (IP) performed on a population of cells to define protein binding sites in the genome using high-throughput sequencing (HTS) (Figure 1.1A). Since its first introduction in 2005, HTS has been a rapidly evolving field, resulting in a broad spectrum of available library preparation protocols and sequencing technologies. Standard HTS platforms include Illumina/Solexa, Roche/454, SOLiD (ABI) and Ion Torrent (Life Technologies) as well as single-molecule approaches by Pacific Biosciences and Oxford Nanopore Technologies. The technologies differ in their sequencing concepts, their throughput and run times, the lengths of the obtained sequence information and the error rates, among others (see [1] for review). Here, we describe the protocol for the Illumina sequencing platform, which is the most widely used. Protocols using other platforms would be similar with slight differences at the library preparation and sequencing steps.
Chromatin preparation The protocol starts with formaldehyde crosslinking of chromatin-bound proteins to DNA, thereby taking a snapshot of how proteins and modifications are distributed along the genome at that point in time. Subsequently, the crosslinked chromatin is sheared until the average DNA fragment length reaches a tight distribution, usually around 200 bp.
Note: Alternatively, ChIP-seq can be performed without crosslinking, which is mostly referred to as native ChIP. In this case, chromatin is digested by micrococcal nuclease (MNase) to reach the desired DNA fragment size. Full digestion leads to ~147 bp fragments corresponding to the length of DNA wound around a single nucleosome. Native ChIP is often used for histone modifications, but also for other factors when crosslinking would mask the epitope recognised by the antibody. However, it is not advisable to use native ChIP for factors that are loosely bound to chromatin, since their contact with DNA might get lost during the protocol.
Immunoprecipitation In the next step, an antibody specific to the protein or modification of interest is used to recognise the corresponding protein-DNA complexes. These complexes are pulled down and enriched using beads binding to the antibody. After a few washing steps that reduce non-specific binding to the beads, proteins are digested, and the extracted co-purified DNA is subjected to library preparation and HTS.
Library preparation Preparing the sequencing library involves repairing the ends of the DNA fragments, and ligating adapters for sequencing. In case of multiplexing, i.e. running more than one sample on the same lane of the flow cell, an indexed adapter is ligated, which allows assigning the sequenced reads to the multiplexed samples after sequencing. The ligation products are purified and PCR-amplified in order to obtain enough material for sequencing. Amplification being a known source of bias, the number of PCR cycles should be kept to a minimum. The library is then loaded onto the flow cell and sequenced.
Sequencing Sequencing of the sheared DNA fragments is always carried out in a 5'-to-3' direction. The forward and reverse adapters are randomly ligated to either end of the double-stranded DNA fragments. Single-end sequencing starts from either the forward or the reverse adapter, while paired-end sequencing is from both ends. The read length is usually shorter than the co-purified DNA fragments, consequently only the fragment ends are sequenced.
Figure 1.1 (A) Schematics of the ChIP-seq experiment. (B) ChIP-seq data analysis workflow.
1.2 IMPROVED DETECTION PROTOCOLS
1.2.1 ChIP-exo
ChIP combined with exonuclease digestion (ChIP-exo) achieves almost nucleotide resolution in defining binding sites [2]. After ligation of the first adapter, E. coli 5'-3' lamda exonuclease is used to trim the co-purified DNA fragments from the 5' end up to a few nucleotides from the crosslink site. The strands are then separated and ligated to the second adapter for sequencing. This means that the reads represent the remainder of the DNA fragment on the 3' side of the crosslink site, with the start of the read lying at or very close to the binding site. This method also reduces the background noise observed in ChIP-seq. ChIP-exo however was designed for SOLiD sequencing platforms, which are not commonly available, and the efficiency of the method is low because it requires two ligation steps.
1.2.2 ChIP-nexus
ChIP experiments with nucleotide resolution through exonuclease, unique barcode and single ligation (ChIP-nexus) is an improvement on ChIP-exo that requires only one ligation step and is adapted for Illumina sequencing platforms [3]. It can identify transcription factor (TF) binding sites in vivo at single nucleotide resolution. In this protocol, the first step after immunoprecipitation is the ligation of an adapter that contains a random barcode (to monitor PCR amplification) and two Illumina sequencing primers separated by a BamHI restriction site. DNA fragments are then digested by the exonuclease that stops at the binding site. DNA is subsequently purified, circularised and digested with BamHI. This results in a product in which the Illumina primers enclose the barcode and the trimmed DNA, that can be amplified and sequenced by single-end sequencing.
1.2.3 CUT&RUN
Cleavage Under Targets and Release Using Nuclease (CUT&RUN) is a technique that is performed in situ and aimed at reducing background noise [4]. In this protocol, the unfixed nuclei are immobilised on magnetic beads and incubated with the antibody specific for the protein or modification of interest and protein A-MNase (pA-MN). pA-MN binds to the antibody and cleaves the DNA around the protein that the antibody is bound to. This means that rather than fragmenting all DNA as in standard ChIP approaches, only protein-bound DNA fragments are excised. Then single nucleosomes or protein-DNA complexes are recovered in the supernatant after centrifugation, and the purified DNA is used for library preparation and high-throughput sequencing.
1.2.4 DamID
DNA adenine methyltransferase identification (DamID) works as an alternative to chromatin immunoprecipitation [5]. We wish to mention it here, as this method can work even if there is no specific antibody available for the TF or nuclear protein of interest. DamID is based on fusing the E. coli adenine methyltransferase to the protein of interest. This fusion protein then creates N6-methyladenine at GATC motifs. The disadvantages of DamID are (i) that it cannot be used for posttranslational modifications such as histone marks, (ii) that the resolution is determined by the GATC frequency in the genome, and (iii) that it does not directly assess occupancy of protein, but just shows that the protein had been present at a given site. Additionally, DamID requires a specialised analysis pipeline.
1.3 CHIP-SEQ DATA ANALYSIS WORKFLOW
ChIP-seq data analysis comprises several steps (Figure 1.1B). The first step after receiving the raw sequence files is performing standard HTS quality control. The aim is to ensure that the sequencing went well, there was no contamination and the library was complex enough (see Chapter 3). Raw reads are then aligned to the reference genome of the studied organism (see Chapter 4). This can be followed by ChIP-seq-specific quality control, that checks for enrichment in the ChIP-seq sample and excludes over-fragmentation (see Chapter 5). The next step is the central analysis in ChIP-seq, which is identifying the genomic regions that are enriched for the factor of interest, referred to as peak calling (see Chapter 6). Once peak regions are defined, ChIP-seq-specific quality control measures that require peak locations can also be assessed (see Chapter 5). Visualisation of the data should be performed at almost every step (see Chapter 7). It is especially important after peak calling to ensure that the predicted peaks accurately capture the binding pattern. The analyses after peak calling depend on the experimental setup and the biological questions. For instance, if there are replicates and/or more than one condition in the experiment, then the next step could be the comparison between samples (see Chapter 8). Biological replicates will give information about reproducibility and intrinsic biological and technical variability in the data (noise). Differential binding analysis addresses which peak regions show significantly different occupancy between two conditions (i.e. differ more than expected from the variation observed between replicates of the same condition). Finally, peak regions or differential peaks can be used in various downstream analysis steps, such as genomic annotation, gene ontology and pathway analyses, motif discovery or integration with additional genomic datasets (see Chapter 9).
1.4 DESIGNING A CHIP-SEQ EXPERIMENT
1.4.1 ChIP-seq controls
In order to confidently identify enriched regions in a ChIP-seq sample, the read distribution needs to be compared to a background distribution to control for potential biases. The optimal control for a ChIP-seq experiment is to sequence the input DNA purified from the sheared chromatin before the antibody incubation step. Other controls can be prepared using different strategies: A mock IP follows all steps of the ChIP-seq protocol but does not use any antibody. An unspecific IP can be performed by using antibodies against a protein that does not bind to chromatin, e.g. immunoglobulin G (IgG). However, both of these approaches lead to small amounts of co-purified DNA with low complexity, which does not reflect the real background distribution. When studying a histone modification, a pan-H3 or pan-H4 antibody that recognises H3 or H4 irrespective of their modifications is a good alternative, as this captures the underlying nucleosome distribution on which the modifications occur.
1.4.2 Sources of bias
Input samples are necessary to control for a number of biases that can cause over-representation of certain regions. Probably the most important source of bias is the non-uniform fragmentation of the chromatin during sonication or digestion. Compact heterochomatic regions are difficult to shear and become under represented compared to open euchromatic regions even in input samples. The effect of chromatin compaction is linear, i.e. the more open the chromatin is the higher its representation in the library [6]. Additionally, the non-uniform fragmentation together with a bias due to PCR amplification will lead to over-representation of GC-rich sequences. Consequently, the background distribution will be positively correlated with the GC content of the genome. This is particularly prevalent in mammalian cells, where euchromatic regions are enriched for CpG islands. Therefore, this should be taken into account when comparing CpG-rich regions with others harbouring fewer CpGs.
Another source of bias arises from the computational processing of the data. During the genomic alignment step, only reads mapping unambiguously to unique positions in the genome are retained (see Chapter 4). This will lead to low coverage in repetitive regions. Finally, in cancer samples and cell lines, the genome can considerably differ from the reference genome sequence. Regions that are deleted in the studied cell line will appear as depleted, while duplicated or amplified regions produce more reads and are seemingly enriched.
To account for these biases, it is crucial to always compare a ChIP sample (or at least a set of replicates) to a control sample, which will be used to control for potential false-positive peaks during the peak calling step (see Chapter 6). If the experimental design involves several conditions, the additional comparison of different samples is useful to cancel out potential biases, including for protocols that do not generate input (e.g. ChIP-exo) (see Chapter 8).
1.4.3 Antibody quality
Since ChIP-seq is an antibody-based immunoprecipitation experiment, its efficiency strongly depends on the quality and specificity...