
eBook - ePub
Handbook of Comparative Pharmacokinetics and Residues of Veterinary Therapeutic Drugs
- 674 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Handbook of Comparative Pharmacokinetics and Residues of Veterinary Therapeutic Drugs
About this book
Handbook of Comparative Pharmacokinetics and Residues of Veterinary Therapeutic Drugs is a unique compilation of comparative pharmacokinetic data for veterinary therapeutic drugs. The book features an excellent introductory chapter on basic veterinary pharmacokinetics and includes pharmacological data taken from hundreds of primary research references. These data are presented in standardized units and are arranged in conveniently organized tables so that comparisons between data can be made easily. Much of the data is new and was taken from articles in which data was not subjected to pharmacokinetic analysis.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
MedicineSubtopic
Veterinary MedicineCHAPTER 1
Principles of Pharmacokinetics
Introduction
Pharmacokinetics is the study of the time course of drug and xenobiotic concentrations in the body. The purpose of this section is to provide an overview of basic pharmacokinetic principles so that the data tabulated in the remainder of the text will be useful. This handbook is a compilation in a single source of as much of the data as is available on the pharmacokinetics and disposition of drugs and xenobiotics encountered in veterinary medicine.
The impetus for compiling this volume was to generate data which would be useful for avoiding drug residues in the edible tissues and products of animal origin.23,30 When drugs are administered to food producing animals either as growth promoters or for therapeutic reasons, a specified period of time must elapse after cessation of drug administration and before slaughter to allow depletion of drug to occur from the animal’s body to levels that are considered “safe” for the human consumer of edible products. These are referred to as the “withdrawal time” for meats and the “milk discard time” for dairy products. In the U.S., the official withdrawal and milk discard times are determined prior to marketing by the Food and Drug Administration based on data submitted by the manufacturer of the product. These times are thus dependent upon the specific formulation of the product as well as on the approved uses and methods of administration, and in most cases, these tests are conducted in healthy animals. The target concentrations which are deemed safe for human consumption are set as “tissue tolerances” by the Food and Drug Administration. When drugs are used in the approved manner in approved species, these legal withdrawal times are generally sufficient. However, occasionally drugs must be used at “extralabel” doses, in non-approved species, or are used inadvertently at excessive dose levels. Sometimes management conditions or disease may also alter drug disposition to the point that the withdrawal time should be lengthened. In other cases, chemicals from the environment may enter the food-producing animal, in which instances, approved withdrawal times and milk discard times do not exist. It is then the task of the veterinarian to estimate a safe withdrawal time or, alternatively, recommend that the animals be disposed of rather then be marketed for human consumption.
The information required to estimate when a drug or chemical is depleted from the animal to safe levels is the realm of the discipline of pharmacokinetics. Pharmacokinetic parameters also allow one to determine tissues in which drugs distribute or accumulate. If data have been collected in both healthy and diseased animals, then one may even be able to predict what effect a disease process may have on drug or chemical depletion times. If these data on the rate and extent of depletion of drugs or chemicals in the body are reduced to a few basic pharmacokinetic parameters, then the information may be extrapolated to individual field situations. Pharmacokinetics also sheds light on the question of interspecies extrapolation. The problem facing the field veterinarian and regulatory personnel is that these data are generally not readily available. In addition, although “raw” depletion data may appear in the published literature, all that is generally tabulated is drug concentrations in different tissues as a function of time after drug administration. Unless these data are analyzed and relevant pharmacokinetic parameters determined, there is little possibility of extrapolating to the field problem at hand. It was thus the purpose of this handbook series to compile this information so that the relevant pharmacokinetic parameters would be readily available to the veterinarian, regulatory personnel, or researcher involved in preventing and mitigating residue problems.
The data sources for this compilation are from the open published literature. Where available, pharmacokinetic parameters calculated by the authors of the primary literature are utilized. However, in many cases, pharmacokinetic parameters were not available as data in the articles consisted solely of tabulations of drug concentration-time data which has not been subjected to mathematical analysis. In most cases, these data are amenable to analysis and a minimal set of pharmacokinetic constants may be calculated if certain assumptions and limitations to interpretation are defined and understood. The procedures used to calculate these data and the assumptions inherent to their derivation are presented in this introductory chapter.
Drug Disposition Overview
In order to understand drug disposition in the animal, the physiologic processes involved must be identified and quantitated. Figure 1 illustrates the processes relevant to a discussion of the disposition of a drug administered by the intravenous (IV), intramuscular (IM), subcutaneous (SC), oral (PO) or topical routes. The reference point for pharmacokinetic analysis is the concentration of free, non-protein-bound drug dissolved in the serum (or plasma), as this is the only body fluid from which samples for drug analysis can be readily and repeatedly collected. For most drugs, serum is in equilibrium with the extracellular fluid of well-perfused tissues; thus, serum drug concentrations generally reflect extracellular fluid drug concentrations, with a few exceptions to be discussed later in this chapter.
Information about the processes of drug distribution and elimination is obtained by carefully monitoring serum drug concentrations over time after an IV injection. Both distribution and elimination tend to remove drug from the systemic circulation, resulting in continually decreasing serum concentrations. If these processes occur at different rates, then the serum drug concentration-time (C-T) profile will reflect this with the steeper slope corresponding to the more rapid process. For most drugs studied, distribution to tissue sites is the more rapid event and is usually the major determinant of the rate of change of drug concentrations immediately after injection. This will continue until equilibrium between serum and tissue concentrations is achieved. Elimination processes will then determine the rate of decline of serum drug concentrations. For certain compounds which show a strong affinity for tissue binding, slow terminal phases in the serum drug C-T profile will be evidence of the release of bound drug from these tissue sequestration sites.
The serum drug C-T profile is thus an integrated picture of the processes affecting drug distribution and elimination in the body. If these processes have significantly different rates, then the C-T profile can be used to analyze them. Figure 2 illustrates this concept with the antibiotic gentamicin in the dog. Over a period of 10 h (inset), two distinct phases related to distribution (a, earliest phase) and elimination (ß, second phase) are evident. Note that during the distribution phase, both distribution and elimination occur simultaneously; however, the more rapid flux of drug to distribution sites predominates. When the distribution and elimination fluxes are in equilibrium, elimination processes then determine the rate of decline in blood concentrations during the second phase. The observed concentration at any time is thus simply a sum of the distribution and elimination contributions. In the so-called elimination phase, the distribution component is negligible. If drug is monitored over a period of 80 h using a more sensitive analytical technique, a third (γ) phase related to tissue binding is detected.
Distribution
Distribution of drug to peripheral tissue sites is dependent upon four factors. These are the physicochemical properties of the drug, the concentration gradient established between the blood and tissue, the ratio of blood flow to tissue mass, and the affinity of the drug for tissue constituents. The physicochemical properties of the drug (pKa, lipid solubility, molecular weight) are most important in determining the propensity of a drug to distribute to a specific tissue site. Tissue distribution is not homogeneous in as much as drugs will have higher affinity for certain tissues depending on regional blood flow, tissue mass, and the biological nature of the tissue. For example, many drugs are rapidly distributed to the liver and kidney because of the large percentage of the cardiac output perfusing these organs. Organs with a high blood flow to mass ratio include the brain, heart, liver, kidney, and endocrine glands; those with an intermediate ratio include muscle and skin; and those with a low ratio, indicative of poor systemic perfusion, are adipose tissue and bone. Lipid-soluble drugs tend to distribute to adipose and brain tissue because of their high lipid content.
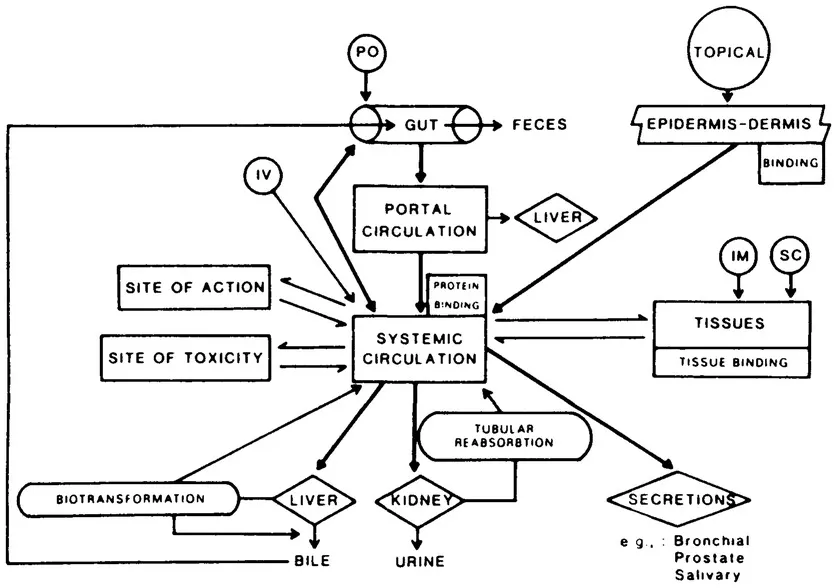
Figure 1 Schematic diagram of the pattern of absorption, distribution, and elimination of a drug. (From Riviere, J.E., in Johnston, The Bristol Veterinary Handbook of Antimicrobial Therapy, 2nd ed., Johnston, D.E., Ed., Veterinary Learning Systems, Lawrenceville, NJ, 1987, 14. With permission.)
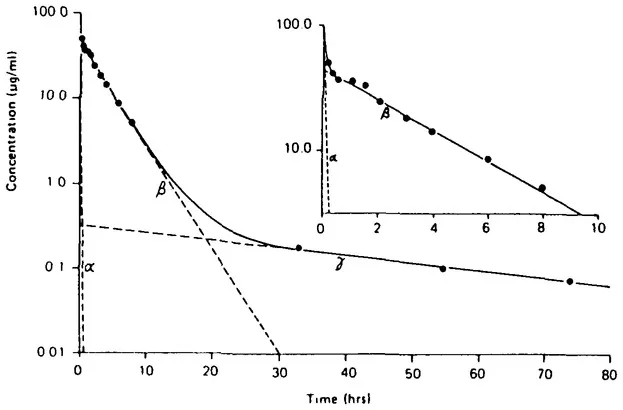
Figure 2 Triphasic pattern of gentamicin elimination in serum of a dog given 10 mg/kg intravenously. (From Riviere, J.E., Compend. Com. Educ. Pracl. Vet., 10, 26, 1988. With permission.)
An important factor which must be considered when discussing drug distribution is the degree of serum protein binding. Only unbound (free) drug is capable of exiting the vascular compartment to exert activity within tissues. The serum protein involved in binding drugs is primarily albumin, although many basic drugs are bound to acid glycoproteins and lipoproteins. Most analytical procedures in common use today measure only total (bound + free) drug concentrations. Serum drug concentrations are generally i...
Table of contents
- Cover
- Title Page
- Copyright Page
- Contents
- The Authors
- Preface
- Acknowledgments
- 1 Principles of Pharmacokinetics
- 2 Orientation to Chapters and Tables
- 3 Central Nervous System Drugs
- 4 Anthelmintic Drugs
- 5 Nonsteroidal Antiinflammatory Drugs
- 6 Narcotics/Opiates
- 7 Hormones
- 8 Cardiovascular And Renal Drugs
- 9 Miscellaneous Drugs
- FDA Approved Tolerance, Action and Safe Levels (ppm)
- Chemical Structures and Physicochemical Properties
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Handbook of Comparative Pharmacokinetics and Residues of Veterinary Therapeutic Drugs by Arthur L. Craigmill in PDF and/or ePUB format, as well as other popular books in Medicine & Veterinary Medicine. We have over 1.5 million books available in our catalogue for you to explore.