
- 148 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Enzymatic Peptide Synthesis
About this book
This book was first published in 1987. Comprehensive overview of enzyme peptide synthesis.
Tools to learn more effectively

Saving Books

Keyword Search

Annotating Text

Listen to it instead
Information
Chapter 1
Introduction
The beginnings of peptide synthetic chemistry* can be traced to 1901 when E. Fischer and E. Fourneau reported on the first systematic synthesis of a dipeptide,1 and further, back to 1882 when T. Curtius unintentionally succeeded in the first in vitro formation of a peptide bond.2 However, the recent advances in peptide synthetic methodology, cumulating in the fully automatic production of polypeptides, commenced only 30 years ago. The starting point of modern peptide synthetic chemistry was marked in 1953 by the chemical synthesis of the nonapeptide hormone oxytocin by du Vigneaud and his collaborators.3 Subsequently, further rapid progress has been stimulated principally by the discovery of an ever-increasing number of biologically active peptides. Whereas a review article published in 1953 and entitled Naturally Occurring Peptides 4 presented the confirmed structures of only six peptides, the compilation of all currently known peptide structures would grow into a herculean task.
The functional versatility of the peptides is astonishing. Their diverse nature encompasses sweeteners and toxins, antibiotics and ionophores, and chemotactic as well as growth factors. Peptides can act both as stimulators and inhibitors of hormone release. They are involved as morphinomimetics in the pain pathways, and in serving as neurotransmitters, they mediate synaptic communication. They consitute enzyme inhibitors, and conversely, they can function as hormonal messengers to activate appropriate target systems.
The isolation and characterization of a hitherto unrecognized peptide almost invariably entails renewed peptide synthetic activities that may aim at a variety of goals. The classical objective of peptide synthesis is the verification or falsification of primary structures elucidated by sequence analyses of bioactive peptides. Furthermore, synthetic peptides, structurally related to their native counterparts, are effective tools to probe the relationship between peptide structure and biological activity. From a pharmacological point of view it is of particular interest that synthetic analogs of peptide hormones may exhibit unique properties such as superpotency, altered biological specificity, and long-lasting activity. Thus, synthetic peptides may be required for therapeutic purposes, particularly if their natural pendants are not easily obtainable in sufficient quantities.
A novel application of peptide synthetic chemistry is in the production of peptides to be used as immunogens in the generation of antisera specific for proteins of which the peptide represents only a part. Although still in its infancy, this rapidly developing technique not only offers exciting possibilities for raising antigenic determinant specific antibodies but it should also deepen our understanding of the basis of antigenicity. Beyond this lies the creation of entirely artificial peptides with unprecedented primary structures. These truly ex arte peptides may be devised either to exhibit putatively nonbiological properties, or to mimic or even enhance commonly known biological activities of their ex natura counterparts. Last but not least, peptide synthetic studies may be performed “simply” for the sake of methodological progress.
*The term “peptide” as coined by E. Fischer generally denotes unbranched chain-like molecules consisting of up to 100 amino acid residues, whereas molecules possessing more than 100 residues are commonly referred to as proteins. Peptides may be further classified into oligo- and polypeptides; the former containing between 2 and 10 and the latter 11 to 100 amino acid units.
At present the most frequently used methods of peptide synthesis are those of a chemical nature. The chemical formation of a peptide bond can, in principle, be reduced to four steps (for more detailed descriptions see References 5 to 10; Figure 1).
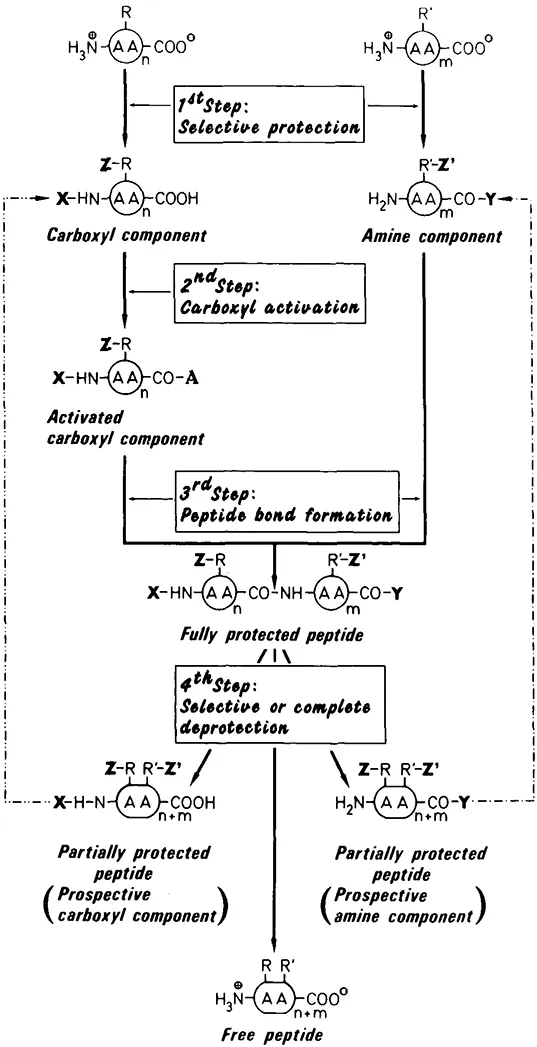
FIGURE 1. Basic scheme of chemical peptide synthesis. AA, amino acid; R,R′ side-chain functionalities; X, α-amine protecting groups; Y, α-carboxyl protecting groups; Z,Z′, side-chain protecting groups; A, activating substituent.
- The main-chain and side-chain functionalities of the educts — amino acids or peptides — which are not to participate in the reaction must be selectively protected.
- The carboxyl component must be activated. (The carboxyl component and the amine component contribute the carbonyl group and the imino group, respectively, to the prospective peptide bond.)
- The peptide bond is formed by coupling the carboxyl component and the amine component via an α-amide linkage.
- The protecting groups must be removed in toto, if the synthesis is completed, or by selective cleavage of the α-amine or α-carboxyl protection, if the synthesis is to be continued.
Originally all synthetic peptides were routinely prepared by conventional solution methods, i.e., the reactants were freely soluble in the reaction media. However, due to B. Merrifield’s ingenious innovation of covalently binding the growing peptide chain to an insoluble polymeric support11 the solid-phase methodology has become increasingly popular over the past 20 years. The most attractive improvement brought about by this procedural simplification is the time-saving and convenient mode of operation and consequently, the prospect of fully automated synthesis.
Numerous peptides have been routinely prepared by solution and solid-phase methods as well as by other techniques such as liquid-phase12 or alternating solid-liquid phase13 procedures and remarkable advances have been made in the art of chemical peptide synthesis. Nevertheless, the considerable shortcomings of these methods still impose an “undiminished challenge”14 upon peptide synthetic chemistry. These limitations arise mainly from the fact that the individual steps of the synthetic pathway are relatively unspecific in nature. Consequently the success of many a synthesis is jeopardized by the appearance of undesired by-products .
To circumvent these problems, an increasing number of organic syntheses is carried out in the presence of enzymes. Due to their stringent specificity, these biocatalysts do not normally permit significant levels of side reactions.
Peptidyltransferase, the enzyme which is responsible for peptide bond formation in vivo, might seem the obvious choice to mediate the enzymatic synthesis of peptides. However, this enzyme is an integral part of the ribosome15 and its activity is dependent upon the presence of additional ribosomal proteins.16 Consequently, it is difficult — if not impossible — to isolate this enzyme in a biologically active form.
In contrast, the proteases, which had previously been considered to be the catalysts of protein biosynthesis,17 are more easily obtainable from their natural sources. Superficially, it may appear incongruous to attempt the synthesis of peptides using proteolytic enzymes. The equilibrium position in a protease-catalyzed reaction is usually far over in the direction of hydrolysis and as a consequence, the reversal of this reaction, i.e., the formation of peptide bonds, represents merely a negligible quantity under physiological conditions. However, the proteases per se cannot be held responsible for this state of affairs. Like other enzymes, they simply accelerate the attainment of equilibrium in a chemical process, whereas the equilibrium point itself, and thus the net synthesis or hydrolysis of the peptide bond, is determined exclusively by thermodynamic factors. Indeed, the proteases do possess the inherent capacity to catalyze both the synthesis and the hydrolysis of a peptide bond, and it is therefore the equilibrium position that actually decides upon the making or breaking of peptide bonds. Consequently, if suitable expedients could be found to shift the equilibrium point of a protease-controlled reaction in favor of the “reverse reaction”, the synthesis of peptide bonds may likely become a considerable quantity.
References
1. Fischer, E. and Fourneau, E., Über einige Derivate des Glykocolls, Ber. Dtsch. Chem. Ges., 34, 2868, 1901.
2. Curtius, T., Über die Einwirkung des Chlorobenzoyl auf Glycocollsilber, J. Prakt. Chem., 24, 239, 1882.
3. du Vigneaud, V., Ressler, C., Swan, J. M., Roberts, C. W., Katsoyannis, P. G., and Gordon, S., The synthesis of a nonapeptide amide with the hormonal activity of oxytocin, J. Am. Chem. Soc., 75, 4879, 1953.
4. Bricas, E., and Fromageot, C., Naturally occurring peptides, Adv. Prot. Chem., 8, 1, 1953.
5. Wünsch, E., Synthese von Peptiden, in Methoden der Organischen Chemie, Vol. 15, Parts 1 and 2, G. Thieme Verlag, Stuttgart, 1974.
6. Lübke, K., Schröder, E., and Kloss, G., Chemie und Biochemie der Aminosären, Peptide und Proteine, Vols. 1 and 2, G. Thieme Verlag, Stuttgart, 1975.
7. Gross, E. and Meienhofer, J., The Peptides: Analysis, Synthesis, Biology, Vols. 1-3, Academic Press, New York, 1979-1981.
8. Jakubke, H.-D. and Jeschkeit, H., Aminosäuren, Peptide, Proteine, Verlag Chemie, Weinheim, 1982.
9. Bodanszky, M., Principles of peptide synthesis, in Reactivity and Structure: Concepts in Organic Chemistry, Vol. 16, Hafner, K., Rees, C. W., Trost, B. M., Lehn, J.-M., v.Ragué-Schleyer, P., and Zahradnik, R., Eds., Springer-Verlag, Berlin, 1984.
10. Bodanszky, M. and Bodanszky, A., The practice of peptide synthesis, in Reactivity and Structure: Concepts in Organic Chemistry, Vol. 21, Hafner, K., Rees, C. W., Trost, B. M., Lehn, J.-M., v.Ragué-Schleyer, P., and Zahradnik, R., Eds., Springer-Verlag, Berlin, 1984.
11. Merrifield, R. B., Solid phase peptide synthesis. I. The synthesis of a tetrapeptide, J. Am. Chem. Soc., 85, 2149, 1963.
12. Mutter, M., Hagenmaier, H., and Bayer, E., Eine neue Methode zur Synthese von Polypeptiden, Angew. Chem., 83, 883, 1971.
13. Frank, H. and Hagenmaier, H., Ein neues Verfahren zur Peptidsynthese: die alternierende Fest-Flüssigphasen Methode, Experientia, 31, 131, 1975.
14. Bodanszky, M., Peptide synthesis: an undiminished challenge, in Peptides, Proc. 5th Am. Peptide Symp., Goodman, M. and Meienhofer, J., Eds., John Wiley and Sons, New York, 1977, 1.
15. Weissbach, H. and Pestka, S., Molecular Mechanisms of Protein Biosynthesis, Academic Press, New York, 1977.
16. Hampl, H., Schulze, H., and Nierhaus, K. H., Ribosomal components from escherichia coli 50S subunits involved in the reconstitution of peptidyltransferase activity, J. Biol. Chem., 256, 2284, 1981.
17. Florkin, M. and Stotz, E. H., Eds., Comprehensive Biochemistry, Vol. 32, Elsevier, Amsterdam, 1977, 307.
Chapter 2
Reversible Zymo-Hydrolysis a Chronology of Enzymatic Peptide Synthesis
The concept of peptide synthesis by reversal of mass action in protease-catalyzed reactions dates back to 1898 when J. H. van’t Hoff suggested that the protease “trypsin”* might posses the inherent capacity to catalyze the synthesis of proteins from degradation products originally generated by its own proteolytic action.2 The rationale behind this idea followed from the applicability of the law of mass action to enzyme-controlled reactions; their reversibility ensuing from the presumed catalytic nature of the process.
The possibility of the participation of hydrolases in not only the degradation but also the assembly of biological macromolecules was first suggested by the phenomenon of the so-called “reversible zymo-hydrolysis”, a term introduced in 1898 by A. C. Hill to designate the maltase-catalyzed synthesis of maltose from glucose.3 Indeed, the earliest reports on glycosidase-, lipase-, and protease-mediated syntheses of glycosides, fats, and peptides were published as early as 18993 and 1901,4,5 respectively, and a further series of studies dealing with enzymatic syntheses of these biomolecules were performed during the following decades (for reviews see References 6 and 7). In contrast, as our present picture of nucleic acid chemistry has evolved only since the early 1950s, it comes as no surprise that the first report of a nuclease-catalyzed formation of oligonucleotides did not appear until 1955.8
As mentioned by J. T. Edsall9 many biochemists, perhaps under the influence of W. Ostwald’s “imperative of energetics” (do not waste any energy, but do exploit it),10 believed that a biochemical process which required free energy to take place could be accomplished with the greatest efficiency by living organisms. As a consequence it was generally taken as granted that the anabolic pathways leading to the synthesis of biological macromolecules were merely reversals of catabolic pathways. This view implicitly suggested that proteins could be prepared by proteolysis in reverse; that is to say, via protease-catalyzed proteosynthesis. The idea of protein biosynthesis by “reversible enzymic hydrolysis” had for some time been considered to b...
Table of contents
- Cover Page
- Title Page
- Copyright Page
- Table of Contents
- Preface
- Acknowledgment
- The Author
- Chapter 1 Introduction
- Chapter 2 Reversible Zymo-Hydrolysis - A Chronology of Enzymatic Peptide Synthesis
- Chapter 3 Thermodynamic Aspects of Peptide Bond Synthesis
- Chapter 4 Formation of Peptide Bonds - Proteases as Activating Systems
- Chapter 5 Proteosynthesis vs. Proteolysis: How to Bias This Antagonism in Favor of Proteosynthesis
- Chapter 6 Advantages of Enzymatic Peptide Synthesis
- Chapter 7 Proteases as Biocatalysts for the Synthesis of Model Peptides
- Chapter 8 Enzymatic Synthesis of Biologically Active Peptides
- Chapter 9 Protease-Catalyzed Oligomerization
- Chapter 10 Protease-Catalyzed Semisynthesis
- Chapter 11 Kinetics of Protease-Controlled Synthesis of Peptide Bonds
- Chapter 12 Proteases as Catalysts in Protecting Group Chemistry
- Chapter 13 Shortcomings and Alternatives
- Chapter 14 Synopsis and Prognosis
- Abbreviations
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Enzymatic Peptide Synthesis by W. Kullman in PDF and/or ePUB format, as well as other popular books in Medicine & Alternative & Complementary Medicine. We have over one million books available in our catalogue for you to explore.