
eBook - ePub
Laboratory Methodology in Biochemistry
Amino Acid Analysis and Protein Sequencing
- 276 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
About this book
Provides information on methodologies and techniques concerning the biochemical laboratory, as well as improvements or advancements made on existing methodologies. Original methodologies for the purification of biological macromolecules and methodologies for metabolic pathways and enzyme kinetics are covered. The application of biochemical and biophysical methodologies for the structural and dynamic characterization of biological macromolecules is considered. The elaboration of automated systems for biochemical research and computer programs for the management and processing of experimental data are both reviewed. Development of instruments and equipment for biochemical research is also presented.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
MedicineSubtopic
Biochemistry in MedicineChapter 1: A Short History Of Protein Sequence Analysis
Table Of Contents
I. Ancient History: 1950—1970
II. The Recent Past: 1970—1985
III. The Modern Era: 1985—
References
I Ancient History: 1950—1970
Amino acid sequence analysis of proteins has progressed through a number of stages since the structure of insulin was determined in 1953 by Frederick Sanger. Dr. Sanger did that analysis at a time when very few of the chemical and instrumental tools of protein chemistry were developed. He had to isolate and characterize more than 150 short peptides from the 51 -residue protein; the anaylsis was extremely labor-intensive and required huge amounts of protein. But it proved once and for all that each protein has a unique amino acid sequence.
Two developments in the mid-1950s made sequence analysis of small (less than Mr 40,000) proteins possible: (1) the development of a quantitative amino acid analyzer by Stanford Moore and William Stein; and (2) Per Edman’s contribution of a sequential chemical-degradation method capable of removing one amino acid at a time cleanly from the amino terminus of a polypeptide. Various enzymatic and chemical cleavage methods were developed to generate peptides 5 to 15 residues in length, and the newly-developing science (art) of column chromatography (mainly Dowex ion exchangers) made it possible to purify the peptides for sequence analyses. A general strategy developed which was used in the 1950s and 1960s to sequence dozens of proteins.
- The protein was cleaved into peptides averaging about 8 to 10 residues in length.
- The peptides were isolated by chromatographic and paper electrophoretic methods.
- A portion of each was acid hydrolyzed to determine its amino acid composition (using the amino acid analyzer), and the rest was subjected to Edman degradation for as many cycles as definitive sequence could be determined (by hand methods that was usually 5 to 15 cycles).
- A different cleavage method was employed on the whole protein and Steps 1 to 3 repeated for that set of peptides.
- Overlapping sequences were aligned to give extended sequence.
- “Holes” in the sequence were filled in by generating yet more sets of small peptides until a complete sequence was obtained.
The above approach was still very labor-intensive and required gram quantities of protein. The size of the protein was limited because large proteins gave more small peptides than the separation methods could resolve.
II The Recent Past: 1970—1985
Automated sequencers became available in 1970. They perform the Edman degradation under rigidly controlled conditions in an inert atmosphere with highly purified reagents. Consequently, the length of readable sequence per degradation increased from an average of 10 or fewer residues to between 30 and 40 (sometimes more). This meant that far fewer peptides had to be isolated and each gave a substantial stretch of sequence, thus reducing the total number of overlapping sequences which were necessary. Roughly a tenfold increase in efficiency, both in terms of labor expended and in protein used, was realized. Continued improvements in peptide isolation techniques and sequencer technology in the 1970s and early 1980s increased the speed of analysis by a further factor of severalfold and vastly reduced the amounts of peptides required in the sequencer reaction chamber (from about 100 nmol in 1970 to 100 pmol in 1985). Much larger proteins could be sequenced by these methods — up to 1,000 residues or so.
In spite of the significant improvements in speed and sensitivity which have been realized over the past two decades, a radically new approach to protein sequence analysis was developed by the mid-1980s (see following section), and no one should contemplate sequencing a whole protein more than 200 residues long by protein sequencer technology any more. Nevertheless, the methods which were developed to accomplish that task are still needed for the new approach and the following paragraphs outline the most important cleavage and peptide separation techniques available to the protein chemist at this time.
Enzymatic Cleavage Methods
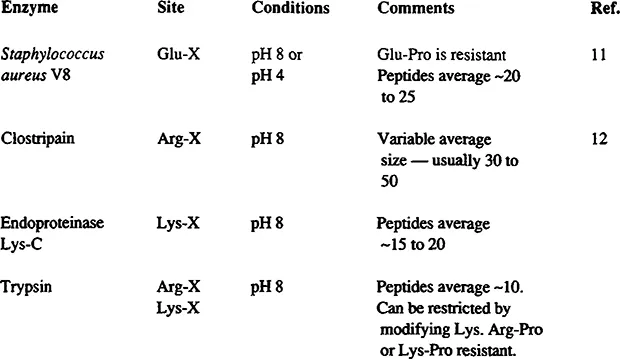
FIGURE 1 All other proteinases are too nonspecific (e.g., Chymotrypsin, pepsin, papain) or too restricted (e.g., thrombin) to be of general use. All four of the enzymes above can be used in urea solution, which increase the rate of digestion and prevents precipitation of partially digested protein. This is crucial for obtaining complete digestion. If >4 M urea is required, small aliquots of proteinase should be added in one hour intervals to compensate for autolysis. The V8 proteinase also cleaves Asp-X bonds if, but only if, phosphate buffers are used, usually not a desirable feature. The reactions are normally run at 5 to 10 mg/ml of protein with 1 to 2% by weight of proteinase. Dropping the pH to about 1 with formic acid terminates the reaction. Upon injection into a reverse-phase HPLC column, the urea, formic acid, and buffer all come out in the breakthrough with peptides emerging in the gradient 13
Application of the automated sequenator to the determination of the total amino acid sequence of a protein required the investigator to generate and purify appropriate fragments of the protein in order to obtain overlapping stretches of sequence covering the whole molecule. Since the sequenator yielded 35 to 50 residues of sequence from a protein fragment, it was most efficient to generate fragments in the 25- to 80-residue size range for the analyses. This required that the cleavage points be rather infrequent in the protein, a situation which also simplified the purification of the fragments by reducing the number of fragments in the digest. The fragmentation procedure also needed to be highly specific for a particular type of bond, so that side reactions (which make fragment purification and identification of certain residues difficult) were not encountered. Finally, the desired bond cleavage needed to proceed in near quantitative yield in order to minimize the number and amounts of fragments generated by incomplete cleavage of a particular bond.
The cyanogen bromide degradation meets these criteria almost perfectly for most proteins. Cyanogen bromide cleaves most Met-X bonds in almost quantitative yield. It is highly specific to Met-X without modifying other residues. Methionyl residues are rare enough in most proteins to yield fragments of ideal size (averaging about 60 residues).
Digestion at arginyl bonds with trypsin can be accomplished by blocking the lysyl residues, preferably with citraconic or succinic anhydride in order to change the charge of the residue from positive to negative. This change makes the denatured protein highly acidic and thus more soluble at pH 8 where the tryptic digestion must be run. Modification with citraconic anhydride has the advantage of reversibility. Thus, merely acidifying the mixture stops the trypsin and removes the blocking groups. Again, this cleavage method is highly quantitative, highly specific, and for many proteins, yields fragments of ideal size. Very often 70 to 80% of a protein sequence could be obtained from sequenator degradations of the cyanogen bromide and arginyl peptides alone. An alternative way to cleave at arginyl residues is to use the enzyme clostripain. It is quite specific for arginyl residues and does not cleave at lysyl residues. The protein to be digested must be dissolved at pH 8, which, for most denatured proteins, requires high concentrations of urea. Clostripain is active in urea solutions, so this is not a serious limitation.
Chemical Cleavage Methods

FIGURE 2 The methods in Figure 1 all produce very large peptides from most proteins. Consequently, the peptides tend to be very difficult to dissolve; they aggregate, and they usually give low yields on HPLC. If sufficient protein is available to use gel filtration procedures, these methods can be very good means of “cracking” a molecule into a small number of pieces; gel filtration should be run in 10 to 20% formic or acetic acids in order to dissolve the peptides and prevent aggregation. 2, 14- 16
Digestion of polypeptides at tryptophanyl residues with o-iodosobenzoic acid generates fragments averaging about 60 residues from most proteins. 1, 2 The yield of cleavage at most Trp- X bonds is close to quantitative. Methionyl and alkylated cysteinyl residues are oxidized to the sulfoxides which can be reduced later with thiol reagents. Free cysteine is oxidized to cystine.
Digestion at glutamyl residues is performed with Staphylococcus aureus protease V8. 3 The protease is active at pH 4.0 where many denatured proteins or peptides are soluble. It is specific for Glu-X bonds provided phosphate is absent (in phosphate, Asp-X bonds are also cleaved). It cleaves quantitatively at most susceptible bonds under proper conditions. Peptides average 15 to 30 residues long.
Specific and quantitative digestion of proteins at cysteinyl residues is also possible 4 and would be exceedingly useful for sequencing if the fragments were not blocked. Since there is no practical way to generate a free amino group from the 2-iminothiazolidine ring formed during the cleavage reaction, this method has limited value for protein-sequencing approaches, but with the new approach employing mass spectral analyses (below), it is once again an important cleavage method.
An enzyme, endoproteinase Lys-C, is available which is specific for Lys-X bonds. It is the preferred way to cleave at those sites. It is active in high concentrations of urea which are usually required to dissolve the denatured peptide at pH 8.
Specific digestion at prolyl residues is also possible. 5 The reaction conditions (sodium metal in anhydrous liquid ammonia) are both dangerous and cumbersome. This digest is rarely employed.
Cleavage can be accomplished at Asn-Gly bonds with hydroxylamine. 6 These bonds are rare in proteins, occurring once or twice in the typical protein. Although the cleavage yield is normally only 70%, the low number of susceptible bonds makes fractionation of the mixture relatively easy. This is an excellent procedure to “crack” a molecule into two or three large pieces and thus reduce the complexity of the cyanogen bromide or arginyl digests.
Likewise, mild acid treatment can give good fragments by cleaving Asp-Pro bonds, another very rare sequence. 7 Strong ac...
Table of contents
- Cover Page
- Title Page
- Copyright Page
- Preface
- The Editor
- Guest Editor
- Advisory Board
- Contributors
- Chapter 1: A Short History Of Protein Sequence Analysis
- Chapter 2: Structural Determination Of Covalently Modified Peptides By Combined Mass Spectrometry And Gas-Phase Microsequencing
- Chapter 3: Amino Acid Analysis Using The Dimethylaminoazobenzene Sulfonyl Chloride (Dabs-C1) Precolumn Derivatization Method
- Chapter 4: Amino Acid Analysis: Protocols, Possibilities, And Pretensions
- Chapter 5: Application Of 4-N,N-Dimethylaminoazobenzene-4’-Isothiocyanate (Dabitc) To The Structure Determination Of Peptides And Proteins*
- Chapter 6: High-Performance Liquid Chromatography (Hplc): A Versatile Tool In Peptide And Protein Chemistry
- 7 Chapter 7: Computational Analysis of Protein Sequencing Data
- 8 Chapter 8: Computers as Tools in Protein Sequencing
- 9 Chapter 9: Mapping Of Ligand Binding Sites, Epitopes, and Posttranslational Modifications On Protein Sequences — Tubulin as an Example
- 10 Chapter 10: ASP-N Endoproteinase, A New Specific Tool in Protein Sequencing
- 11 Chapter 11: Enzymatic Digestion of Proteins and HPLC Peptide Isolation in the Subnanomole Range
- 12 Chapter 12: HPLC and Hormone Purification — Application to Atrial Natriuretic Factor
- 13 Chapter 13: Electroblotting: A Method For Protein Purification For NH2-Terminal And Internal Microsequencing
- 14 Chapter 14: Methods For Determination Of Protein Sequences By Fast Atom Bombardment Mass Spectrometry
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Laboratory Methodology in Biochemistry by C. Fini,Carlo Fini in PDF and/or ePUB format, as well as other popular books in Medicine & Biochemistry in Medicine. We have over 1.5 million books available in our catalogue for you to explore.