Volumes in the Proven Synthetic Methods Series address the concerns many chemists have regarding irreproducibility of synthetic protocols, lack of identification and characterization data for new compounds, and inflated yields reported in chemical communications—trends that have recently become a serious problem.
Featuring contributions from world-renowned experts and overseen by a highly respected series editor, Carbohydrate Chemistry: Proven Synthetic Methods, Volume 4 compiles reliable synthetic methods and protocols for the preparation of intermediates for carbohydrate synthesis or other uses in the glycosciences.
Exploring carbohydrate chemistry from both the academic and industrial points of view, this unique resource brings together useful information into one convenient reference. The series is unique among other synthetic literature in the carbohydrate field in that, to ensure reproducibility, an independent checker has verified the experimental parts involved by repeating the protocols or using the methods.
The book includes new or more detailed versions of previously published protocols as well as those published in not readily available journals. The essential characteristics of the protocols presented are reliability, updated characterization data for newly synthesized substances and the expectation of wide utility in the carbohydrate field. The protocols presented will be of wide use to a broad range of readers in the carbohydrate field and the life sciences, including undergraduates taking carbohydrate workshops.

eBook - ePub
Carbohydrate Chemistry
Proven Synthetic Methods, Volume 4
- 369 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Carbohydrate Chemistry
Proven Synthetic Methods, Volume 4
About this book
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
MedicineSubtopic
Biochemistry in MedicineSection I
Synthetic Intermediates
11 Synthesis of 4-Nitrophenyl β-D-galactofuranoside
A Useful Substrate for β-D-Galactofuranosidases Studies
Carla Marino*, Santiago Poklepovich Caridea and Rosa M. de Lederkremer
Universidad de Buenos Aires, Pabellón II, Ciudad Universitaria
Sydney Villaume†
University of Namur (UNamur)
CONTENTS
Experimental
General Methods
4-Nitrophenyl 2,3,5,6-tetra-O-benzoyl-β-D-galactofuranoside
4-Nitrophenyl β-D-galactofuranoside
Acknowledgments
References
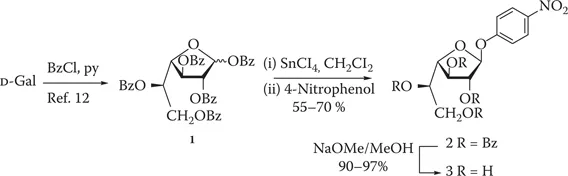
β-D-Galactofuranosyl units (β-D-Galf) are constituents of microorganisms, some of them pathogenic, such as Mycobacteria, the trypanosomatids Trypanosoma cruzi and Leishmania,1 and fungi such as Aspergillus fumigatus.2 Since Galf has never been found in mammals, its biosynthesis and metabolism are good targets for chemotherapeutic strategies. In some species, the degradation of Galf-containing glycoconjugates is promoted by extracellular β-D-galactofuranosidases. For example, Penicillium and Apergillius species,3 Helminthosporium sacchari,4 and Trichoderma harzianum5 produce exo β-D-galactofuranosidases (EC 3.2.1.146).
First studies of β-D-galactofuranosidases involved the use of methyl β-D-galactofuranoside as substrate and the tedious measurement of the reducing sugar released by the enzyme.4,6 The availability of the chromogenic substrate 4-nitrophenyl β-D-galactofuranoside (3), first described in our laboratory7 and later reported by Cousin et al.,8 significantly simplified this task. Since then, compound 3 has been extensively used for galactofuranosidase studies in P. fellutanum,9 and Aspergillius spp.8 Compound 3 has also been used as a substrate for chemoenzymatic syntheses.10,11 It used to be commercially available (Merck) but it was later discontinued. In our case, the starting compound was per-O-benzoyl-α,β-D-Galf (1),12 obtained in a single step from D-Gal, and the glycosylation was promoted by p-toluenesulfonic acid.7 The other laboratory started from per-O-acetyl-β-D-Galf, which required three steps for its synthesis, and the glycosylating agent was SnCl4,8 which proved to be very efficient for the synthesis of different galactofuranosides.13 In this chapter, we describe the synthesis of 3 by SnCl4-promoted glycosylation of easily available 1,12 followed by de-O-acylation.
EXPERIMENTAL
General Methods
Thin-layer chromatography (TLC) was performed on 0.2 mm silica gel 60 F254 (Merck) aluminum-supported plates. Detection was effected by UV light and by charring with 10% (v/v) H2SO4 in EtOH. Column chromatography was performed on silica gel 60 (230–400 mesh, Merck). The proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were recorded with a Bruker AM 500 spectrometer at 500 MHz (1H) and 125.8 MHz (13C). Assignments were supported by homonuclear correlation spectroscopy (COSY) and heteronuclear single quantum coherence (HCQC) experiments. Optical rotations were measured with a Perkin-Elmer 343 polarimeter, with a path length of 1 dm. Melting points were determined with a Fisher-Johns apparatus and are uncorrected. CH2Cl2 was distilled from P2O5 and stored over 4 Å MS. MeOH was dried by refluxing over magnesium turnings and a little iodine, distilled, and stored over 4 Å MS.
Sodium methoxide was prepared by carefully reacting a sub-stoichiometric amount of sodium (∼0.020–0.025 g) with dried methanol (5 mL).
4-Nitrophenyl 2,3,5,6-tetra-O-benzoyl-β-D-galactofuranoside (2)
A 50 mL, single-neck round-bottom flask equipped with a rubber septum was charged with 1,2,3,5,6-penta-O-benzoyl-α,β-D-galactofuranose (1, 1.05 g, 1.50 mmol),* dry CH2Cl2 (10.0 mL), and 4 Å MS (0.1 g). The reaction vessel was flushed with argon and, with magnetic stirring and external cooling in an ice-water bath, SnCl4 (0.25 mL, 2.1 mmol, 99% purity)* was added, followed after 10 min by addition of 4-nitrophenol (0.25 g, 1.8 mmol)†. The cooling was removed, and the reaction mixture was allowed to reach room temperature.
After 3 h of stirring, TLC showed consumption of the starting material (Rf ∼0.6 both anomers, 9:1 toluene–EtOAc) and presence of main product (Rf 0.7). Excess 4-nitrophenol is also observed by TLC. The mixture was filtered, diluted with CH2Cl2 (50 mL), and successively washed with NaHCO3 (2.5%, 2 × 20 mL)‡ and sat aq NaCl (20 mL). The organic layer was dried (Na2SO4), filtered, and concentrated. The syrup obtained (1.95 g) was chromatographed (49:1 toluene–EtOAc) to afford syrupy 2 (654 mg, 61%), [α]D −11 (c 1, CHCl3).7 1H NMR (500 MHz, CDCl3) δ 8.16–7.15 (H-aromatic), 6.10 (m, 1 H, H-5), 6.07 (s, J < 0.5 Hz, 1 H, H-1), 5.78 (s, J < 0.5 Hz, 1 H, H-2), 5.77 (d, J 4.8 Hz, 1 H, H-3), 4.80 (apparent t, J 4.3 Hz, 1 H, H-4), 4.75 (m, 2 H, H-6, 6′); 13C NMR (50.3 MHz, CDCl3) δ 165.9, 165.7, 165.6, 165.4 (PhCO), 142.7–116.5 (C-aromatic), 103.8 (C-1), 83.1 (C-4), 81.9 (C-2), 77.3 (C-3), 69.9 (C-5), 63.0 (C-6). Anal. Calc. for C40H31NO12: C, 66.94; H, 4.35; N, 1.95. Found: C, 67.17; H, 4.36; N, 1.77.7
4-Nitrophenyl β-D-galactofuranoside (3)
A dry 100 mL, single-neck round-bottom flask with a magnetic stirring bar and a rubber septum was charged with 2 (0.5 g, 0.69 mmol) and dried in vacuo for 1 h at room temperature. Anhydrous CH2Cl2 (8 mL) was added and with stirring and external cooling in an ice-water bath 0.2 M methanolic NaOMe in anhydrous MeOH (2.5 mL) was added in one portion. After 30 min, TLC analysis (7:1:2 PrOH-28% aq NH3) showed complete conversion of the starting material into a more polar product (Rf 0.5), faster-moving than a galactose standard (Rf 0.2). The solution was concentrated to a small volu...
Table of contents
- Cover Page
- Title Page
- Copyright Page
- Dedication Page
- Contents
- Foreword
- Introduction
- Editors
- Series Editor
- Contributors
- SECTION I Synthetic Methods
- SECTION II Synthetic Intermediates
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Carbohydrate Chemistry by Christian Vogel, Paul Murphy, Christian Vogel,Paul Murphy in PDF and/or ePUB format, as well as other popular books in Medicine & Biochemistry in Medicine. We have over 1.5 million books available in our catalogue for you to explore.