
- 328 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
About this book
Presents authoritative state-of-the-art discussions of the key issues pertinent to transdermal drug delivery, examining those topics necessary to enable a critical evaluation of a drug candidate's potential to be delivered across the skin; from physical chemistry and assessment of drug permeability to available enhancement technolgies, to regulator
Tools to learn more effectively

Saving Books

Keyword Search

Annotating Text

Listen to it instead
Information
Topic
MedicineSubtopic
Dermatology1
Feasibility Assessment in Topical a1nd Transdermal Delivery: Mathematical Models and In Vitro Studies
I. INTRODUCTION
Fortunately for us, the skin has evolved into an extremely efficient barrier, which prevents both excessive water loss from the body and the ingress of xenobiotics. It enables us to withstand a considerable range of environmental challenges. The reasons for this are manifold and may be summarized simply for the purposes of this chapter.
The outer layer of the skin, the stratum corneum, forms the rate-controlling barrier for diffusion for almost all compounds (Fig. 1).
It is composed of dead, flattened, keratin-rich cells, the corneocytes. These dense cells are surrounded by a complex mixture of intercellular lipids. They comprise ceramides, free fatty acids, cholesterol, and cholesterol sulphate. Their most important feature is that they are structured into ordered bilayer arrays. The predominant diffusional path for a molecule crossing the stratum corneum appears to be intercellular (1). The diffusant therefore follows a tortuous route and has to cross, sequentially, and repeatedly, a number of hydrophilic and lipophilic domains. It is not surprising, therefore, that the lipid-water partitioning characteristics of the permeant is a dominant determinant of its penetration or that mathematical models developed to predict percutaneous absorption include a term to describe partitioning. Fick’s laws of diffusion describe the diffusional step, and although the skin is a heterogeneous membrane, simple solutions can often be applied. This will be appreciated later when these laws and their limitations are described.
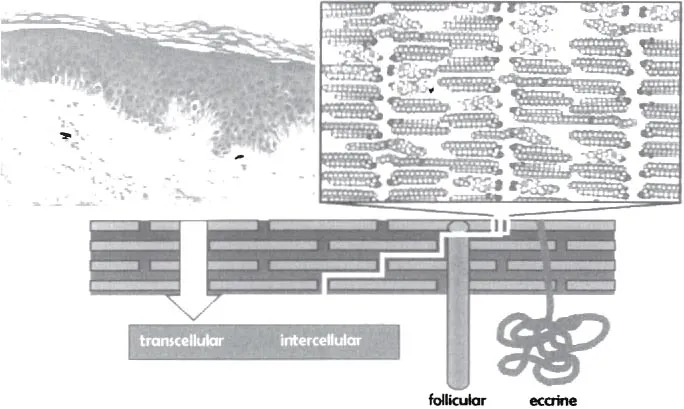
Figure 1 A schematic representation of the skin. Top left is a photomicrograph of the skin, clearly showing the stratum corneum, viable epidermis, and underlying dermis. Top right is a representation of the structured lipids found in the intercellular channels. The bottom illustration shows the different routes that a diffusant can take in its passage through the stratum corneum, although the predominant one is usually intercellular.
There are a number of reasons why it is important to predict the absorption rate. For dermal drug delivery, it is desirable to choose a drug structure that has the best possible chance of arriving at the site of action. It should be remembered that compounds such as the corticosteroids have a bioavailability of only a few percent (2). If drugs were chosen with more favorable partition and diffusion characteristics, this bioavailability could be improved considerably. An accurate and descriptive mathematical model would enable this rational choice and facilitate the design of novel topical agents. For transdermal delivery, sufficient drug must permeate the various strata of the skin so as to build up a plasma concentration, which elicits a systemic effect. This route has a number of attractions, and an accurate and predictive model would be invaluable in the selection and evolution of appropriate transdermal drug candidates. Equally, there are also chemicals, the absorption of which in significant amounts is clearly undesirable. Compounds such as pesticides are obvious examples, but there are other materials, present perhaps as formulation excipients, that could also be detrimental. An appropriate mathematical model would allow a reliable risk assessment to be made before in vivo evaluations are conducted.
There are different considerations to be taken into account depending on whether the drug is to be delivered for local action or for systemic action. Since this book concerns primarily transdermal delivery, the major emphasis will be how to ensure the transport of drug through the skin into the underlying dermal vasculature and hence the systemic circulation. For a drug to be administered transdermally, it has to be very potent, as it is unlikely that more than a few tens of milligrams per day can be delivered. To a first approximation, feasibility can be assessed from the daily dose. But, as will be seen, even for a compound like nitroglycerine, which has ideal physicochemical properties for transdermal delivery from a reasonable patch area, no more than 40 to 50 mg per day can be delivered.
In some ways, it is more difficult to assess the feasibility of topical drug delivery, as the levels required in the skin for therapeutic effect are usually unknown. For transdermal delivery, there is a well-documented and determinable end point, the plasma level required for efficacious therapy. Advances in noninvasive monitoring and microdialysis can be helpful in determining the target skin concentration for topical therapy, but data are limited, and the reliability of the methodologies involved is still in question, as the techniques remain in very much a developmental stage.
Validated mathematical models represent an economically advantageous approach for the assessment of skin permeation, and their use is recommended before full-blown in vitro and in vivo experiments are conducted. The purpose of this chapter is to examine the limitations of mathematical modeling and to consider appropriate in vitro models prior to full clinical testing.
II. FICK’S LAWS OF DIFFUSION
Considering that the skin is such a heterogeneous membrane, it is surprising that simple diffusion laws can be used to describe the percutaneous absorption process (3). Since transdermal delivery involves the application of a device over a long period of time, it is generally assumed that steady-state conditions have been reached and that the most relevant law of diffusion is therefore Fick’s first law. The second law describes non-steady state diffusion and can be used to analyze the rates of release from matrix type transdermal patches, to evaluate the lag phase prior to the establishment of steady-state conditions, and to describe concentration profiles across the skin as they evolve towards linearity.
The most quoted form of Fick’s first law of diffusion describes steady-state diffusion through a membrane:
(1)
where J is the flux per unit area, K is the stratum corneum-formulation partition coefficient of the drug, and D is its diffusion coefficient in the stratum corneum of path length h; co is the concentration of drug applied to the skin surface, and ci is the concentration inside the skin. In most practical situations, co ⨠ ci and Eq. (1) simplifies to
(2)
where kp (= DK/h) is the permeability coefficient, which has units of velocity (often quoted as cm h−1), i.e., it is a heterogeneous rate constant and encodes both partition and diffusional characteristics. The input rate of the drug into the systemic circulation, from a patch of area A, is therefore given by the product
(3)
The output or elimination rate from the systemic circulation equals the clearance (Cl) multiplied by the plasma concentration at steady state (cp,ss)
(4)
Hence Eqs. (3) and (4) may be combined to predict the drug’s plasma concentration following transdermal delivery:
(5)
The plasma concentration achieved therefore depends directly on the area of the device, the skin permeability, and the applied concentration and is inversely related to the drug’s clearance (4).
For a given drug, the clearance and the target plasma level are likely to be known, so to examine the feasibility of delivery, one needs the drug’s skin permeability and its solubility, as this will give an indication of the maximum concentration that can be applied. These parameters can be estimated from basic physicochemical properties, which are typically measured during preformulation.
III. ESTIMATION OF PERMEABILITY COEFFICIENT FROM SIMPLE PHYSICOCHEMICAL PARAMETERS
Over the years, a database of skin permeability values has been consolidated (e.g., Ref. 5). To avoid confusion and to simplify interpretation, permeabilities from aqueous solutions alone are considered. The use of skin permeabilities from other vehicles is complicated in that most other solvents are capable of altering the barrier function of skin. Analysis of the existing database enabled Potts and Guy to formulate an empirical relationship between kp and two simple characteristics of the permeant: the octanol-water partition coefficient (Koct) and the molecular weight (MW) (6).
(6)
If log Koct has not been measured, or is not available, it can easily be predicted by commercially available algorithms. The latter are increasingly more reliable and accurate.
Equation 6 shows that, as log Koct increases, the permeability also increases, whereas the greater the molecular weight the smaller is kp. Permeants that are best absorbed through the skin are therefore small and lipophilic. This analysis often gives rise to confusion, as it must be realized that it is not the permeability coefficient alone that determines the efficiency of topical and transdermal delivery. Rather, it is the flux across the skin, which is the product of the permeability coefficient and the drug concentration in the vehicle. As the dataset under discussion concerns percutaneous transport from aqueous solution, the maximum achievable flux is therefore kp multiplied by the aqueous solubility (Sw). Often, this value is also known from preformulation studies; if not, it can also be estimated from equations such as (7):
(7)
where ΔSf is the entropy of fusion of the permeant (which can also be estimated), mp is its melting point, and T is the temperature at which the solubility is required. It is immediately apparent that (a) as Koct increases, the aqueous solubility decreases, and (b) the lower the melting point, the higher the solubility (i.e., a reflection of the role of intermolecular forces on solubility). Solubilities can also be estimated using commercially available software; typically, the algorithms employed require knowledge of the melting point and the octanol-water partition coefficient. Since the maximum flux is the product of kp and Sw, simply increasing the drug’s Koct results...
Table of contents
- Cover
- Title Page
- Advisory
- Series
- Halftitle Page
- Copyright Page
- Preface
- Contents
- Contributors
- 1. Feasibility Assessment in Topical and Transdennal Delivery: Mathematical Models and In Vitro Studies
- 2. Evaluating the Transdennal Permeability of Chemicals
- 3. Skin Absorption Databases and Predictive Equations
- 4. Partitioning of Chemicals into Skin: Results and Predictions
- 5. Iontophoresis: Applications in Drug Delivery and Noninvasive Monitoring
- 6. Skin Electroporation for Transdennal and Topical Drug Delivery
- 7. Sonophoresis: Ultrasound-Enhanced Transdennal Drug Delivery
- 8. Metabolic Approach to Transdennal Drug Delivery
- 9. The Application of Supersaturated Systems to Percutaneous Drug Delivery
- 10. Minimally Invasive Systems for Transdennal Drug Delivery
- 11. Transdennal Drug Delivery System Regulatory Issues
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Transdermal Drug Delivery Systems by Jonathan Hadgraft in PDF and/or ePUB format, as well as other popular books in Medicine & Dermatology. We have over one million books available in our catalogue for you to explore.