
- 768 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Drug-Drug Interactions
About this book
Authored by renowned leaders in the field, this comprehensive volume covers all aspects of drug-drug interactions, including preclinical, clinical, toxicological, and regulatory perspectives.Thoroughly updated, this second edition reflects the significant advances and includes extensive new material on:key interplay between transporters and enzymes
Tools to learn more effectively

Saving Books

Keyword Search

Annotating Text

Listen to it instead
Information
Topic
MedicineSubtopic
Pharmacology
Introducing Pharmacokinetic and Pharmacodynamic Concepts
University of Manchester, Manchester, U.K.
I. SETTING THE SCENE
All effective drugs have the potential for producing both benefits and risks associated with desired and undesired effects. The particular response to a drug by a patient is driven in one way or another by the concentration of that drug, and sometimes its metabolites, at the effect sites within the body. Accordingly, it is useful to partition the relationship between drug administration and response into two phases, a pharmacokinetic phase, which relates drug administration to concentrations within the body produced over time, and a pharmacodynamic phase, which relates response (desired and undesired) produced to concentration. In so doing, we can better understand why patients vary in their response to drugs, which includes genetics, age, disease, and the presence of other drugs.
Patients often receive several drugs at the same time. Some diseases, such as cancer and AIDS, demand the need for combination therapy, which works better than can be achieved with any one of the drugs alone. In other cases, the patient is suffering from several conditions, each of which is being treated with one or more drugs. Given this situation and the many potential sites for interaction that exist within the body, it is not surprising that an interaction may occur between them, whereby either the pharmacokinetics or the pharmacodynamics of one drug is altered by another. More often than not, however, the interaction is of no clinical significance, because the response of most systems within the body is graded, with the intensity of response varying continuously with the concentration of the compound producing it. Only when the magnitude of change in response is large enough will an interaction become of clinical significance, which in turn varies with the drug. For a drug with a narrow therapeutic window, only a small change in response may precipitate a clinically significant interaction, whereas for a drug with a wide margin of safety, large changes in, say, its pharmacokinetics will have no clinical consequence. Also, it is well to keep in mind that some interactions are intentional, being designed for benefit, as often arises in combination therapy. Clearly, those of concern are the unintentional ones, which lead to either ineffective therapy through antagonism or lower concentrations of the affected drug or, more worryingly, excessive toxicity, which sometimes is so severe as to limit the use of the offending drug or, if it produces fatality, result in its removal from the market.
This chapter lays down the conceptual framework for understanding the quantitative and temporal aspects of drug-drug interactions, hereafter called drug interactions for simplicity. Emphasis is placed primarily on the pharmacokinetic aspects, partly because pharmacokinetic interactions are the most common cause of undesirable and, to date, unpredictable interactions and also because most of this book is devoted almost exclusively to this aspect and indeed to one of its major components, drug metabolism. Some pharmacodynamic aspects are also covered, however, for there are many similarities between pharmacokinetic and pharmacodynamic interactions at the molecular level and because ultimately one has to place a pharmacokinetic interaction into a pharmacodynamic perspective to appreciate the likely therapeutic impact (1, 2, 3, 4, 5).
II. BASIC ELEMENTS OF PHARMACOKINETICS
As depicted in Figure 1, it is useful to divide pharmacokinetic processes in vivo broadly into two parts, absorption and disposition. Absorption, which applies to all sites of administration other than direct injection into the bloodstream, comprises all processes between drug administration and appearance in circulating blood. Bioavailability is a measure of the extent to which a drug is absorbed. Disposition comprises both the distribution of a drug into tissues within the body and its elimination, itself divided into metabolism and excretion of unchanged drug. Disposition is characterized independently following intravenous administration, when absorption is not involved.
Increasingly, aspects of potential drug interactions are being studied in vitro not only with the aim of providing a mechanistic understanding but also with the hope that the findings can be used to predict quantitatively events in vivo, and thereby avoid or limit undesired clinical interactions. To achieve this aim, we need a holistic approach whereby individual processes are nested within a whole body frame—that is, constructs (models) that allow us to explore the impact, for example, of inhibition or induction of a particular metabolic pathway on, say, the concentration-time profile of a drug in the circulating plasma or blood, which delivers the drug to all parts of the body, including sites of action and elimination. This approach also allows us to better interpret the underlying events occurring in vivo following a drug interaction. To appreciate this last statement, consider the events shown in Figures 2 and 3 and the corresponding summary data given in Table 1.
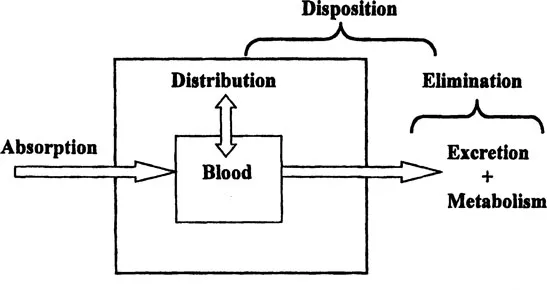
Figure 1 Schematic representation of processes comprising the pharmacokinetics of a compound. Here terms are defined with respect to measurement in blood or plasma. Absorption comprises all events between drug administration and appearance at the site of measurement. Distribution is the reversible transfer of the drug from and to other parts of the body. Elimination is the irreversible loss of the drug either as unchanged compound (excretion) or by metabolism. Disposition is the movement of the drug out of blood by distribution and elimination.
In Figure 2, pretreatment with the antibiotic rifampin shortened the half-life and decreased the area under the plasma concentration-time curve (AUC) profile, but not materially the peak concentration, of the oral anticoagulant warfarin, whether given intravenously or orally. In contrast, pretreatment with the sedative-hypnotic pentobarbital reduced both the peak concentration and AUC of the antihypertensive agent alprenolol following oral administ...
Table of contents
- Cover
- Half Title
- Title Page
- Copyright Page
- Table of Contents
- Preface
- Contributors
- 1. Introducing Pharmacokinetic and Pharmacodynamic Concepts
- 2. In Vitro Enzyme Kinetics Applied to Drug-Metabolizing Enzymes
- 3. Human Cytochromes P450 and Their Role in Metabolism-Based Drug-Drug Interactions
- 4. UDP-Glucuronosyltransferases
- 5. Drug-Drug Interactions Involving the Membrane Transport Process
- 6. In Vitro Models for Studying Induction of Cytochrome P450 Enzymes
- 7. In Vitro Approaches for Studying the Inhibition of Drug-Metabolizing Enzymes and Identifying the Drug-Metabolizing Enzymes Responsible for the Metabolism of Drugs (Reaction Phenotyping) with Emphasis on Cytochrome P450
- 8. The Role of P-Glycoprotein in Drug Disposition: Significance to Drug Development
- 9. Cytochrome P450 Protein Modeling and Ligand Docking
- 10. Role of the Gut Mucosa in Metabolically Based Drug-Drug Interactions
- 11. Mechanism-Based Inhibition of Human Cytochromes P450: In Vitro Kinetics and In Vitro–In Vivo Correlations
- 12. Transporter-Mediated Drug Interactions: Molecular Mechanisms and Clinical Implications
- 13. Metabolism and Transport Drug Interaction Database: A Web-Based Research and Analysis Tool
- 14. In Vivo Probes for Studying Induction and Inhibition of Cytochrome P450 Enzymes in Humans
- 15. Drug-Drug Interactions: Clinical Perspective
- 16. An Integrated Approach to Assessing Drug-Drug Interactions: A Regulatory Perspective
- 17. Drug-Drug Interactions: Toxicological Perspectives
- 18. Drug-Drug Interactions: Marketing Perspectives
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Drug-Drug Interactions by A. David Rodrigues in PDF and/or ePUB format, as well as other popular books in Medicine & Pharmacology. We have over one million books available in our catalogue for you to explore.