
- 232 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
About this book
Recent Advances in Ophthalmology focuses on developments in ophthalmology, including therapy, keratotomy, phototoxicity, and retinopathy. The compilation first offers information on the therapy of herpes simplex, anterior segment surgery, and use of viscous and viscoelastic substances in ophthalmology. Topics include systemic antiviral agents and ocular disease; influence of new instruments and technology; criteria for the selection of viscous or viscoelastic material; and indications for the use of viscous substances. The text then ponders on radial keratotomy, intraocular lenses, and treatment of congenital cataracts. The publication takes a look at ocular phototoxicity and vitreous surgery, as well as proliferative diabetic retinopathy, posterior penetrating trauma, effects of non-ionizing radiation on biological systems, body temperature and light damage, and retinal photodamage. The text also touches on the management of diabetic retinopathy; treatment of malignant melanomas of the posterior uvea; and macular disease with serious retinal detachment. The compilation is highly recommended for ophthalmologists and readers interested in the developments in ophthalmology.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
MedicineSubtopic
Surgery & Surgical Medicine1
Therapy of herpes simplex
H.E. Kaufman
Publisher Summary
The chapter discusses the therapy of herpes simplex. For antiviral drugs to be effective and to interact with DNA polymerase, they must be present in the cell in the triphosphate form. The first phosphate is added to the molecule either by cellular thymidine kinase or by the virus-encoded enzyme. The virus-encoded thymidine kinase is much more active than the cellular enzyme. Two additional phosphates are rapidly added by cellular enzymes and the compound is then able to interact with the DNA polymerase. There may be some direct inhibition of DNA synthesis through abnormal attachments and binding to the DNA polymerase; however, in general, it seems that the nucleosides with normal sugars and abnormal bases act by incorporation and false coding. One of the antivirals to be developed for ophthalmic use was adenine arabinoside. This arabinose sugar probably acts largely as a chain terminator and is an effective topical antiviral. It is as insoluble as idoxuridine.
INHIBITORS OF NUCLEIC ACID SYNTHESIS
Before 1961, there was no really effective treatment for virus infections of the cornea, and there were no specific antiviral drugs. Ocular herpetic infections were treated by means of epithelial debridement, which appeared to have some benefit. However, frequently caustic agents were used (Gundersen, 1936) that were capable of denaturing proteins and causing stromal scarring. It was never clear that this treatment was not worse than the disease. Antimetabolites had been considered as possible agents for use in virus chemotherapy, but virtually all that had been tried were drugs that inhibited the cellular production of nucleosides needed for DNA synthesis. These drugs were never effective because the herpes virus DNA polymerase had a greater affinity for the nucleosides than the cellular DNA polymerase; therefore it was virtually impossible to stop virus multiplication without totally destroying all normal cells as well.
Idoxuridine
Since the DNA polymerase of the virus was known to be specifically coded and was known to have different affinities, compared to the cellular DNA polymerase (Herrmann, 1961), it seemed reasonable that effective virus chemotherapy would have to involve a drug that worked specifically on the viral DNA polymerase, either by inhibiting its activity or by being incorporated itself into a false DNA chain, which would no longer code proper genetic material and therefore would not reproduce, so that new virus would not be made. We first tested idoxuridine because it was very similar to thymidine chemically (Kaufman, 1962) (Table 1.1). The van der Waal’s radius of the iodine group was similar to that of the methyl group of thymidine, and it was felt that this similarity would allow idoxuridine to interfere with the uptake of thymidine in the synthetic process. The chloro- and bromo-deoxyuridines appeared to be more toxic than the iodinated form of the molecule.
Table 1.1
Possible sites of inhibition of DNA synthesis by antiviral compounds

Iodo-deoxyuridine was synthesised by William Prusoff, of the Department of Pharmacology at Yale University, approximately three years before its first use as an ocular antiviral agent (Prusoff, 1959). The compound was developed originally as an anticancer drug but was relatively ineffective. From the biochemical studies that were carried out at that time, it was known that idoxuridine directly inhibited DNA polymerase and was incorporated into a false DNA. For this reason, and because of its lesser toxicity, this compound was tested as a topical antiviral, and it was found to be effective. A series of double-blind studies confirmed the activity of idoxuridine, but a similar drug, 5-fluorouracil, was found to inhibit the synthesis of thymidine, rather than its uptake, and was not an effective in vivo antiviral agent.
The testing of antiviral drugs was a new area. With further work, it became clear that in animal and clinical studies, the use of complex scoring systems that included redness of the eye, iritis, etc. (Draize et al, 1944) clouded the precise measurement of drug efficacy and made the prediction of drug potency difficult. The results were more precisely predictive when we concentrated on the size of the corneal ulcer alone and the improvement in corneal healing during drug administration.
One of the problems with idoxuridine in its original form was its relative lack of solubility. In part because of this, it was necessary initially to administer the drug every hour during the day and every two hours at night; later, a longer-lasting ointment was used at night. However, clearly, idoxuridine was effective and clinically useful in the treatment of ocular herpes (Kaufman et al, 1962).
Mechanism of action
For antiviral drugs to be effective and to interact with DNA polymerase, they must be present in the cell in the triphosphate form. The first phosphate is added to the molecule either by cellular thymidine kinase or by the virus-encoded enzyme; the virus-encoded thymidine kinase is much more active than the cellular enzyme. Two additional phosphates are rapidly added by cellular enzymes and the compound is then able to interact with the DNA polymerase.
All of the metabolite inhibitors have multiple sites of activity, and it is not always certain which is the true rate-limiting mechanism of action. In spite of this, we can hypothesise a mechanism of action for these drugs. DNA is a helix with a backbone of sugar molecules hooked to each other with 3-,5-bonds, forming a long chain. Attached to the backbone are the bases: adenine, thymine, guanine, and uracil. These bases are read by chemical processes to produce messenger RNA that provides the code for protein synthesis. The base pattern is also involved in the duplication of DNA. The bases themselves are not essential to the structure of the backbone although theoretically, they could stereochemically hinder the formation of the helix.
It appears that the nucleoside analogues that have normal sugar moieties and abnormal purine or pyrimidine substituents for some of the bases, probably are incorporated in the formation of DNA, but the mistakes in the base pattern code for products do not permit the replication of the virus. There may be some direct inhibition of DNA synthesis through abnormal attachments and binding to the DNA polymerase, but in general it seems that the nucleosides with normal sugars and abnormal bases act by incorporation and false coding. Those nucleosides with abnormal sugars—especially abnormalities involving the three or five positions on the carbon chain, which are necessary to link up the sugars to make the DNA chain — probably act largely as chain terminators. They are picked up by the DNA polymerase and one bond forms to a normal sugar molecule of the normal DNA chain, but the chain cannot be continued because the sugar is too abnormal to sustain a 3–5 bonding and permit the synthesis of the backbone of the chain. Thus small pieces of DNA are synthesised and the synthesis of a complete DNA chain is interrupted. This explanation is somewhat oversimplified, and some of the mechanisms are still somewhat uncertain, but it does provide enough of an overview to explain how antiviral drugs exert their effects.
Adenine arabinoside
The next topical antiviral to be developed for ophthalmic use was adenine arabinoside (Underwood, 1962). This arabinose sugar probably acts largely as a chain terminator and is an effective topical antiviral. It is as insoluble as idoxuridine. There has been some suggestion that adenine arabinoside is slightly less toxic than idoxuridine on a chronic basis, but there is no good evidence for this (Dresner & Seamans, 1975). Evidence from double blind studies (Pavan-Langston & Dohlman, 1972) indicate that adenine arabinoside has approximately the same potency as idoxuridine (Table 1.2) and also about the same short term topical toxicity (Dresner & Seamans, 1975). It is also clear that neither adenine arabinoside nor idoxuridine is effective as a primary therapeutic agent in the treatment of stromal disease and iritis.
Table 1.2
Efficacy of idoxuridine and adenine arabinoside
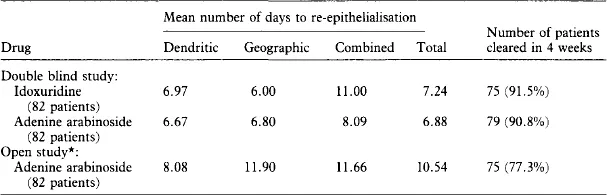
*Generally the severity of the disease and the duration of symptoms were greater in patients entered in the open study
(adapted from Dresner & Seamans, 1975).
Trifluorothymidine
Trifluorothymidine, like idoxuridine, was synthesised originally as an anti-cancer drug by Gottschling & Heidelberger (1963). However, trifluorothymidine has several unique properties. It is extremely potent, perhaps because it has a dual mechanism of action; it inhibits the de novo synthesis of thymidine (as a thymidylic synthetase inhibitor), and it also acts directly as a substitute thymidine analogue on DNA polymerase (Heidelberger et al, 1962). It is extremely soluble, so that high concentrations can be obtained in drop form, and ointments are not necessary (Kaufman & Heidelberger, 1964). The drops are usually given between five and eight times a day in the treatment of dendritic ulcer (Wellings et al, 1972), and approximately 97% of patients are cured within two weeks. Resistance to trifluorothymidine occurs, but it is very rare, possibly because of the multiple sites of activity. In the few cases where resistance occurs, the older, less effective drugs can be used. Trifluorothymidine is the most effective topical antiviral now available for the treatment of ocular epithelial herpes, and because of the low incidence of viral resistance, is probably the drug of choice for ordinary topical use.
Out work, as well as studies by others, suggests that trifluorothymidine is also the most potent drug now available for counteracting the steroid effect, when steroids are used as a primary therapeutic agent. Epithelial toxicity from trifluorothymidine is seen infrequently, unless the drug is misused and continued at high frequency for a long time. The drops should not be applied on a frequent dosage schedule for a period of many weeks; when the early high frequency applications are reduced rapidly after about five to seven days, corneal toxicity is rare.
All of the topical antivirals can produce epithelial toxicity. Idoxuridine may appear to be somewhat more toxic, since allergy may play a part in this reaction. The eye may become red, the epithelium keratinised, and punctal occlusion also occurs with herpes infections alone, so it is not certain that idoxuridine therapy is implicated in this problem. All of these antiviral drugs — idoxuridine, adenine arabinoside, and trifluorothymidine—may produce typical allergenic reactions, and all of them ma...
Table of contents
- Cover image
- Title page
- Table of Contents
- Inside Front Cover
- Copyright
- Preface
- Contributors
- Chapter 1: Therapy of herpes simplex
- Chapter 2: Anterior segment surgery
- Chapter 3: The use of viscous and viscoelastic substances in ophthalmology
- Chapter 4: Radial keratotomy
- Chapter 5: Intraocular lenses
- Chapter 6: Treatment of congenital cataracts
- Chapter 7: Ocular phototoxicity
- Chapter 8: Vitreous surgery
- Chapter 9: The management of diabetic retinopathy
- Chapter 10: Current treatment of malignant melanomas of the posterior uvea
- Chapter 11: Macular disease with serious retinal detachment
- Chapter 12: Glaucoma
- Chapter 13: Laser trabeculoplasty
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Recent Advances in Ophthalmology by S. I. Davidson,F. T. Fraunfelder in PDF and/or ePUB format, as well as other popular books in Medicine & Surgery & Surgical Medicine. We have over 1.5 million books available in our catalogue for you to explore.