
eBook - ePub
Frontiers in Computational Chemistry: Volume 2
Computer Applications for Drug Design and Biomolecular Systems
- 444 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Frontiers in Computational Chemistry: Volume 2
Computer Applications for Drug Design and Biomolecular Systems
About this book
Frontiers in Computational Chemistry, originally published by Bentham and now distributed by Elsevier, presents the latest research findings and methods in the diverse field of computational chemistry, focusing on molecular modeling techniques used in drug discovery and the drug development process. This includes computer-aided molecular design, drug discovery and development, lead generation, lead optimization, database management, computer and molecular graphics, and the development of new computational methods or efficient algorithms for the simulation of chemical phenomena including analyses of biological activity. In Volume 2, the authors continue the compendium with nine additional perspectives in the application of computational methods towards drug design. This volume covers an array of subjects from modern hardware advances that accelerate new antibacterial peptide identification, electronic structure methods that explain how singlet oxygen damages DNA, to QSAR model validation, the application of DFT and DFRT methods on understanding the action of nitrogen mustards, the design of novel prodrugs using molecular mechanics and molecular orbital methods, computational simulations of lipid bilayers, high throughput screening methods, and more.
- Brings together a wide range of research into a single collection to help researchers keep up with new methods
- Uniquely focuses on computational chemistry approaches that can accelerate drug design
- Makes a solid connection between experiment and computation, and the novel application of computational methods in the fields of biology, chemistry, biochemistry, physics, and biophysics
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
Physical SciencesSubtopic
PharmacologyChapter 1
The Use of Dedicated Processors to Accelerate the Identification of Novel Antibacterial Peptides
Gabriel del Rio1,*; Miguel Arias-Estrada1; Carlos Polanco González2,3 1 Computer Science Department, Instituto Nacional de Astrofísica, Óptica y Electrónica, Puebla, Puebla, México
2 Departamento de Matemáticas, Facultad de Ciencias, Universidad Nacional Autónoma de México. Cd.Universitaria, 04510 México, D.F. México
3 Subdirección de Epidemiología Hospitalaria y Control de Calidad de la Atención Médica, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Vasco de Quiroga 15, Col. Sección XVI 14000 D.F. México
* Department of Biochemistry and Structural Biology, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, México DF, México [email protected]
2 Departamento de Matemáticas, Facultad de Ciencias, Universidad Nacional Autónoma de México. Cd.Universitaria, 04510 México, D.F. México
3 Subdirección de Epidemiología Hospitalaria y Control de Calidad de la Atención Médica, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Vasco de Quiroga 15, Col. Sección XVI 14000 D.F. México
* Department of Biochemistry and Structural Biology, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, México DF, México [email protected]
Abstract:
In the past decades, the procedure to identify novel antibiotic compounds has been motivated by the heuristic discovery of the antibiotic penicillin by Fleming in 1929. Since then, researches have been isolating compounds from very wide range of living forms with the hope of repeating Fleming’s story. Yet, the rate of discovery of new pharmaceutical compounds has reached a plateau in the last decade and this has promoted the use of alternative approaches to identify antibiotic compounds. One of these approaches uses the accumulated information on pharmaceutical compounds to predict new ones using high-performance computers. Such approach brings up the possibility to screen for millions of compounds in computer simulations. The better predictors though use sophisticated algorithms that take up significant amount of computer time, reducing the number of compounds to analyze and the likelihood to identify potential antibiotic compounds. At the same time, the appearance of computer processors that may be tailored to perform specific tasks by the end of the past century provided a tool to accelerate high-performance computations. The current review focuses on the use of these dedicated processor devices, particularly Field Programmable Gate Arrays and Graphic Processing Units, to identify new antibacterial peptides. For that end, we review some of the common computational methods used to identify antibacterial peptides and highlight the difficulties and advantages these algorithms present to be coded into FPGA/GPU computational devices. We discuss the potential of reaching supercomputing performance on FPGA/GPU, and the approaches for parallelism on these platforms.
Keywords
Antibacterial peptides
FPGA
GPU
high-performance computations
parallelism
QSAR
supercomputing
Introduction
The discovery of salvarsan (arsfenamine) in 1901 by Paul Erlich facilitated the treatment of syphilis; Erlich proposed the idea of “magic bullets” to explain the way this synthetic compound was able to kill the bacteria associated to the disease: a magic bullet (salvarsan) traversing along the body in search of its target (bacteria) without damaging any other tissue [1]. This discovery promoted the idea of synthesizing target-specific compounds as a way to find novel antibiotics. In 1929 penicillin was accidentally discovered by Flemming [2] and provided an example of an effective antibiotic whose target was unknown at the time of discovery; this in turn promoted the development of phenotype screening methods aimed to identify antibiotics by their function and not by specific target. These two strategies are still in use; for instance, in the period from 1999 to 2008, 28 out of 45 first-in-class new molecules tested by the FDA were discovered by phenotype screening methods [3]. Each of these strategies has advantages and disadvantages that have been recently reviewed [4, 5] and are out of the scope of this review. For any of these strategies, knowledge about the molecular mechanism of action of antibiotics is a desirable feature for any drug to be used and that implies knowing the target of action; however, many antibiotics approved by the FDA are poly-pharmacologic (i.e., act on multiple targets) [6] and such feature troublesome the synthesis of new pharmacologic compounds based on specific targets [7] because that implies the synthesis of large molecules that tend to be non-permeable to cells and not easy to synthesize.
The recent recognition that polypharmacological compounds are among the most effective antibiotics may explain the relatively small number of new pharmaceutical compounds approved by the FDA despite the increasing amount of resources invested [8]. This has led to a shift in the last decade in the research and development strategy of the pharmaceutical industry: focus on strategic therapeutic areas and outsourcing with Universities. Among the areas gaining interest in pharmaceutical industries are new treatments for bacterial infections. This trend constitutes an impulse to explore new strategies to identify new antibacterial compounds, especially those obtained from biological means, also referred to as biologies. We review the results of a new promising strategy aimed to identify new antibacterial compounds combining the knowledge gained from the traditional target-based or phenotype-based strategies with computer sciences and technology.
Antibacterial Peptides
Antibacterial peptides (APs) are produced by many different organisms and have the ability to arrest cell growth (bacteriostatic) or to kill bacteria (bactericidal) [9]. Similar to other antibacterial compounds [10], APs act upon different targets in bacterial cells [11]: the cell wall, DNA replication machinery and the ribosome; furthermore, these peptides not only act on the bacterial cell, but also are able to elicit an immune response from the host as part of the innate immune response [12]. Furthermore, some APs also have antiviral, antiparasitic and antifungal activity that had led to use some APs (e.g., gramicidin S and polymyxin B) to treat infections [13]; yet some features of natural APs must first be improved before they can be used as therapeutics, including: the high cost of large-scale production, stability to proteases, unspecific toxicity against eukaryotic cells, and potential development of immunological reactions [14]. Among these, the cost of production and toxicity against eukaryotic cells seem feasible to be improved simultaneously by producing linear peptides in biological systems [15] and alter the physical properties of AP to achieve selectivity towards bacteria [16]. In the current review, we analyze the strategies that have been used to identify selective antibacterial peptides mainly focus on computational approaches.
The most abundant APs are cationic antibacterial peptides or CAPs, which are relatively short (12-100 residues) and amphiphilic; despite their similar physical properties, CAPs share very little sequence similarity and fold into four main classes: amphiphilic peptides with two to four β-strands, amphipathic α-helices, loop structures, and extended structures [12]. The amphipathic α-helical peptides are linear and are suitable for large-scale production since these do not require any disulfide bond to adopt a functional structure [17].
The mechanism of action for any CAP is accepted to require the interaction of the peptide with the bacterial membrane, but the basis of their action differs according to their final target of action [18, 19] (see Fig. 1): CAPs would approach be stabilized around membrane due to their rich composition of Arginine residues, which have the ability to interact with both lipid and water; once in contact with the membrane, CAPs may disrupt it or pass through it to find its target [20].
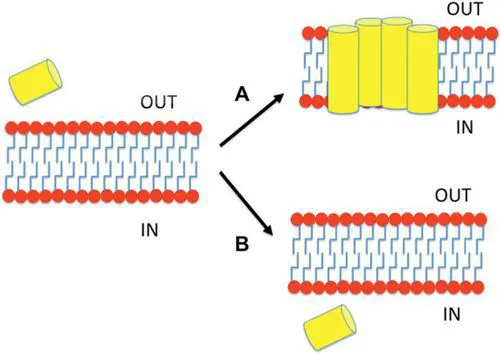
Figure 1 Mechanisms of action of CAPs. CAPs may interact with membrane and either destabilize the lipid membrane (A) or insert into the membrane in a non-disruptive manner (B). A CAP is represented in the image as a yellow cylinder and the lipid membrane as red circles with blue tails.
In any case, a desirable feature of CAPs is the ability to discriminate bacterial from mammalian cells; such peptides are referred here to as Selective CAPs, or SCAPs. Different computational approaches have been reported that aimed to identify novel antibacterial peptides [21-25].
Considering the extremely large number of possible peptide sequences, it is expected that systematic computational screenings may be performed on small peptides (less than 30 amino acid residues). Noticeable, the most active antibacterial peptides can be found in peptides of very different lengths (see Fig. 2; the figure shows only peptides with MIC against Eschericia coli < = 10 μM). Interestingly, the majority of these peptides have been isolated from the animal kingdom, which should display preferential antibiotic activity against bacteria and few if any toxicity against animal cells, that is, these may be part of the SCAPs.
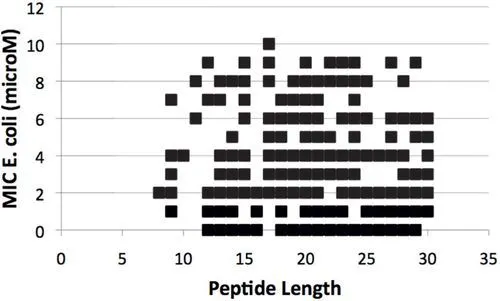
Figure 2 Size-Activiy relationship of APs. The reported activity against Escherichia coli by every antibacterial peptide reported in the YADAMP with 30 or less amino acids in length, with Minimum Inhibitory Conc...
Table of contents
- Cover image
- Title page
- Table of Contents
- Copyright
- Preface
- List of Contributors
- Chapter 1: The Use of Dedicated Processors to Accelerate the Identification of Novel Antibacterial Peptides
- Chapter 2: Computational Chemistry for Photosensitizer Design and Investigation of DNA Damage
- Chapter 3: How to Judge Predictive Quality of Classification and Regression Based QSAR Models?
- Chapter 4: Density Functional Studies of Bis-alkylating Nitrogen Mustards
- Chapter 5: From Conventional Prodrugs to Prodrugs Designed by Molecular Orbital Methods
- Chapter 6: Structural and Vibrational Investigation on a Benzoxazin Derivative with Potential Antibacterial Activity
- Chapter 7: First Principles Computational Biochemistry with deMon2k
- Chapter 8: Recent Advances in Computational Simulations of Lipid Bilayer Based Molecular Systems
- Chapter 9: Data Quality Assurance and Statistical Analysis of High Throughput Screenings for Drug Discovery
- Subject Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Frontiers in Computational Chemistry: Volume 2 by Zaheer Ul-Haq,Jeffry D. Madura in PDF and/or ePUB format, as well as other popular books in Physical Sciences & Pharmacology. We have over 1.5 million books available in our catalogue for you to explore.