
- 484 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
About this book
Written by leading cell biologists and curated by Cell Press editors, reviews in the Cell Press Reviews: Core Concepts in Cell Biology publication informs, inspires, and connects cell biologists at all stages in their careers with timely, comprehensive insight into the most recent exciting developments across cell biology and hot topics within core areas of the field including:
- Signaling mechanisms and membrane biology
- Cytoskeletal self-organization and cell polarity
- Organelle dynamics and biogenesis
- Morphogenesis and cell motility
- Chromatin and genome organization in nuclear function
Contributions come from leading voices in cell biology, who are defining the future of their field, including:- Tom Misteli, National Cancer Institute - Galit Lahav, Harvard Medical School - Scott D. Emr, Cornell University - David G. Drubin, University of California, Berkeley - Tom Rapoport, Harvard Medical School - Anthony A. Hyman, Max Planck Institute of Molecular and Cell Biology, Dresden
This publication is part of the Cell Press Reviews series, which features reviews published in Cell Press primary research and Trends reviews journals.
- Provides timely, comprehensive coverage across a broad range of cell biological topics
- Offers foundational knowledge and expert insights to students and others new to the field
- Features reviews from leaders in cell biology research and discussion of future directions for the field
- Includes articles originally published in Cell, Current Biology, Developmental Cell, and Trends in Cell Biology
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Subtopic
BiologyIndex
Biological SciencesChapter 1
The Cell Biology of Genomes
Bringing the Double Helix to Life
Tom Misteli1,∗, 1National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA, ∗Correspondence: [email protected]
The recent ability to routinely probe genome function at a global scale has revolutionized our view of genomes. One of the most important realizations from these approaches is that the functional output of genomes is affected by the nuclear environment in which they exist. Integration of sequence information with molecular and cellular features of the genome promises a fuller understanding of genome function.
Keywords
genome; stochastic; epigenetic; genome organization
Acknowledgments
Due to space limitations, mostly review articles were cited. Work in the author’s laboratory is supported by the Intramural Research Program of the National Institutes of Health (NIH), NCI, Center for Cancer Research.
Cell, Vol. 152, No. 6, March 14, 2013 © 2013 Elsevier Inc.
http://dx.doi.org/10.1016/j.cell.2013.02.048
Summary
The recent ability to routinely probe genome function at a global scale has revolutionized our view of genomes. One of the most important realizations from these approaches is that the functional output of genomes is affected by the nuclear environment in which they exist. Integration of sequence information with molecular and cellular features of the genome promises a fuller understanding of genome function.
Introduction
It was a moment of scientific amazement in 1953 when Watson and Crick revealed the structure of DNA. The magnificence of the double helix and its elegant simplicity were awe inspiring. But more than just being beautiful, the double helix immediately paved the way forward; its structure implied fundamental biological processes such as semiconservative replication and the notion that chemical changes in its composition may alter heritable traits. The linear structure of DNA laid the foundation for the concept that a string of chemical entities could encode the information that determines the very essence of every living organism. The beauty of the double helix was the promise that, if the sequence of bases in the genome could be mapped and decoded, the genetic information that underlies all living organisms would be revealed and the secret of biological systems would be unlocked.
The idea of linearly encoded genetic information has been spectacularly successful, culminating in the recent development of powerful high-throughput sequencing methods that now allow the routine reading of entire genomes. The conceptual elegance of the genome is that the information contained in the DNA sequence is absolute. The order of bases can be determined by sequencing, and the result is always unequivocal. The ability to decipher and accurately predict the behavior of genome sequences was appealing to the early molecular biologists, has given rise to the discipline of molecular genetics, and has catalyzed the reductionist thinking that has driven and dominated the field of molecular biology since its inception.
But the apparent simplicity and deterministic nature of genomes can be deceptive. One of the most important lessons learned from our ability to exhaustively sequence DNA and to probe genome behavior at a global scale by mapping chromatin properties and expression profiling is that the sequence is only the first step in genome function. In intact living cells and organisms, the functional output of genomes is modulated, and the hard-wired information contained in the sequence is often amplified or suppressed. While mutations are an extreme case of genome modulation, most commonly occurring changes in genome function are more subtle and consist of fluctuations in gene expression, temporary silencing, or temporary activation of genes. Although not caused by mutations, these genome activity changes are functionally important.
Several mechanisms modulate genome function (Figure 1). At the transcription level, the limited availability of components of the transcription machinery at specific sites in the genome influences the short-term behavior of genes and may make their expression stochastic. Epigenetic modifications are capable of overriding genetically encoded information via chemical modification of chromatin. Similarly, changes in higher-order chromatin organization and gene positioning within the nucleus alter functional properties of genome regions.
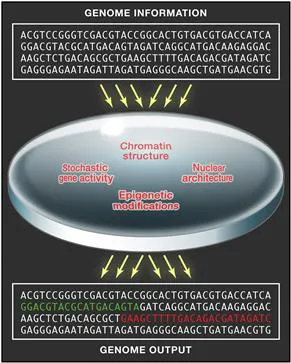
FIGURE 1 From Primary Sequence to Genome Output
The hard-wired primary information contained in the genome sequence is modulated at short or long timescales by several molecular and cellular events. Modulation may lead to activation (green) or silencing (red) of genome regions.
The hard-wired primary information contained in the genome sequence is modulated at short or long timescales by several molecular and cellular events. Modulation may lead to activation (green) or silencing (red) of genome regions.
The existence of mechanisms that modulate the output of genomes makes it clear that a true understanding of genome function requires integration of what we have learned about genome sequence with what we are still discovering about how genomes are modified and how they are organized in vivo in the cell nucleus.
The Stochastic Genome
The genome is what defines an organism and an individual cell. It is therefore tempting to assume that identical genomes behave identically in a population of cells. We now know that this is not the case. Individual, genetically identical cells can behave very differently even in the same physiological environment. It is rare to find a truly homogeneous population of cells even under controlled laboratory conditions, as anyone who has tried to make a cell line stably expressing a transgene knows. Much of the variability in biological behavior between individual cells comes from stochastic activity of genes (Raj and van Oudenaarden, 2008).
Genes are by definition low-copy-number entities, as each typically only exists in two copies in the cell. Similarly, many transcription factors are present in relatively low numbers in the cell nucleus. The low copy number of genes and transcription factors makes gene expression inherently prone to stochastic effects (Raj and van Oudenaarden, 2008). Numerous observations make it clear that gene expression is stochastic in vivo. For example, dose-dependent increases in gene expression after treatment of cell populations with stimulating ligands, such as hormones, are often brought about by high expression of target genes in a relatively small number of cells in the population rather than by a uniform increase in the activity in all cells. Stochastic gene behavior is most evident in single-cell imaging approaches, and mapping by fluorescence in situ hybridization of multiple genes, which according to population-based PCR analysis are active in a given cell population, shows that only a few cells transcribe all “constitutively active” genes at any given time. Most cells only express a subset of genes, and the combinations vary considerably between individual cells. These observations suggest that many genes blink on and off and are expressed in bursts rather than in a continuous fashion (Larson et al., 2009).
The molecular basis for stochastic gene expression is unknown. There are several candidate mechanisms, all of which are related to genome or nuclear organization. Most genes require some degree of chromatin remodeling for activity, which is thought to make regulatory regions accessible to the transcription machinery. Several observations suggest that chromatin remodeling contributes to the stochastic bursting of gene expression. Maybe most compelling is the finding that genes located near each other on the same chromosome show correlated blinking behavior, indicating that a local chromosome property, such as chromatin structure, drives stochastic behavior (Becskei et al., 2005). Furthermore, altering chromatin, for example by deletion of chromatin remodeling machinery, affects stochastic variability in yeast. It can be envisioned that the stochastic behavior of genes is caused by the requirement for cyclical opening of chromatin regions. Open chromatin has a limited persistence time, and maintaining chromatin in an open state requires the cyclical action of chromatin remodelers. Whether an “active” gene is transcribed at any given time may thus depend on the transient condensation status of its chromatin at a particular moment.
A second mechanism to impose nonuniform stochastic genome activity may be the local availability of the transcription machinery at a gene. Although transcription factors are able to relatively freely diffuse through the nuclear space, and in this way effectively scan the genome for binding sites, their availability and functionality at a given local site may undergo significant temporal fluctuations (Misteli, 2001). The local availability of transcription complexes may affect transcription frequency positive or negatively. On the one hand, it is possible that relatively stable preinitiation complexes persist on a given gene, where they may support multiple rounds of transcription and in this way boost initiation frequency. On the other hand, assembly of the full polymerase is a stochastic and relatively inefficient event itself. In order for a functional polymerase complex to assemble, individual transcription machinery components associate with chromatin in a step-wise fashion, and formation of the mature polymerase complex involves multiple partially assembled intermediates, many of which are unstable and disintegrate before a functionally competent complex is formed (Misteli, 2001). The inefficiency of polymerase assembly may create stochasticity at an individual locus.
A further contributor to stochastic gene expression may be the organization of transcription events in transcription factories. These hubs of transcription consist of accumulations of transcription factors to which multiple genes, often located on distinct chromosomes, are recruited (Edelman and Fraser, 2012). Typically only a few hundred such transcription factories are observed in a mammalian cell nucleus. It is possible that some genes need to physically relocate from nucleoplasmic locations to transcription factories. A nominally “active” gene locus that is not associated with a transcription factory may thus be stochastically silent. The relatively low number of transcription sites makes them a limiting factor in the transcription process and thus a potential mediator of stochastic gene expression.
Epigenetics—and When Epigenetics is not Epigenetics
Stochastic effects modulate genome output on short timescales. A mechanism to modulate the hardwired information of genomes on longer timescales is via epigenetics. The Greek-derived “Epi” means “over” or “above,” and epigenetic effects are defined as heritable changes in genome activity caused by mechanisms other than changes in DNA sequence. Epigenetic events are mediated by chemical modifications of DNA or core histones in complex patterns by methylation, acetylation, ubiquitination, phosphorylation, etc. These modifications alter gene expression by changing the chromatin surface and in this way affect the binding of regulatory factors. Well-established examples of such effects include binding of the DNA-methylation-dependent binding of the MeCP2 protein or the binding of PHD-domain-containing proteins to trimethylated histone H3 tails. Prominent biological effects based on epigenetic regulation are phenotypic differences between homozygous twins or imprinted genes that are expressed from only one allele in a diploid organism.
A central tenet in the definition of epigenetic regulation is that its effects are heritable, i.e., transmittable over generations. In fact, the concept of epigenetics was inspired by epidemiological findings that nutrient availability in preadolescents during the 19th century Swedish famine determined life expectance of their grandchildren. The epidemiological studies have recently been complemented by controlled laboratory studies in mice (Rando, 2012), and they have been extended to the molecular level by the findings that loss of the histone H3K4-trimethylation prolongs lifespan in C. elegans in a heritable fashion for several generations (Greer et al., 2011).
A complicating aspect of epigenetics is that the same modifications that mediate heritable epigenetic regulation may also bring about nonheritable transient modulations of the genome. In fact, the term “epigenetic” is nowadays often used in a very cavalier manner to refer to any biological effect, heritable or not, that is affected by histone modifications. Even if they are not heritable, histone modifications are biologically relevant modulators of genome function. The system of histone modifications is in many ways akin to the mechanisms by which signal transduction pathways work (Schreiber and Bernstein, 2002). Just as in signal transduction pathways, posttranslational modifications on histone tails create binding sites that are then recognized by adaptor or reader proteins, which in turn elicit downstream effects such as activation of kinases in the case of signaling cascades or recruitment of transcription factors in the case of histone modifications. In further analogy to the reversible events in signaling pathways, histone modifications can be altered or erased by modifying enzymes. Such transient and reversible modulatory effects of histone modifications have been implicated in every step of gene expression, starting from chromatin remodeling to recruitment of transcription machinery and even to downstream events that were thought to be chromatin independent, such as alternative pre-mRNA splicing (Luco et al., 2011). It is often difficult to determine heritability of these histone modification effects, and it therefore remains unclear how many of them are truly epigenetic. Regardless, DNA and histone modifications are an obvious source of modulation of the information contained in the genome sequence.
Genome Organization as a Modulator of Genome Function
Genomes of course do not exist as linear, naked DNA in the cell nucleus but are organized into higher-order chromatin fibers, chromatin domains, and chromosomes. Many correlations between genome organization and activity have been made—most prominently, the findings that transcriptionally active genes are generally located in decondensed chromatin and that transcriptionally repressed genome regions are often found at the nuclear periphery. These observations point to the possibility that the spatial organization of the genome modulates its functional output.
But in considering the relationship of genome structure with its function, we are faced with a perpetual chicken-and-egg problem. Does structure drive function, or is structure merely a reflection of function? Much of the thinking on this topic has been guided by observations on individual genes. How representative these were for the genome as a whole has been a confounding concern. Recent unbiased genome-wide analysis of structure/function relationships has validated the tight link between structure and function. Large-scale analysis of chromatin structure, histone modifications, and expression profiles shows ...
Table of contents
- Cover image
- Title page
- Table of Contents
- Copyright
- About Cell Press
- Contributors
- Preface
- Chapter 1. The Cell Biology of Genomes: Bringing the Double Helix to Life
- Chapter 2. Condensin, Chromatin Crossbarring and Chromosome Condensation
- Chapter 3. Nuclear Positioning
- Chapter 4. Evolution and Function of the Mitotic Checkpoint
- Chapter 5. Cell Division Orientation in Animals
- Chapter 6. Cell Polarity: Mechanochemical Patterning
- Chapter 7. Encoding and Decoding Cellular Information through Signaling Dynamics
- Chapter 8. Directed Cytoskeleton Self-Organization
- Chapter 9. Mitochondria in Apoptosis: Bcl-2 Family Members and Mitochondrial Dynamics
- Chapter 10. Golgi Membrane Dynamics and Lipid Metabolism
- Chapter 11. Weaving the Web of ER Tubules
- Chapter 12. The ESCRT Pathway
- Chapter 13. Mechanisms of Autophagosome Biogenesis
- Chapter 14. Organelle Growth Control through Limiting Pools of Cytoplasmic Components
- Chapter 15. Curvature, Lipid Packing, and Electrostatics of Membrane Organelles: Defining Cellular Territories in Determining Specificity
- Chapter 16. Clathrin-Mediated Endocytosis in Budding Yeast
- Chapter 17. Life at the Leading Edge
- Chapter 18. Use the Force: Membrane Tension as an Organizer of Cell Shape and Motility
- Chapter 19. Thinking Outside the Cell: How Cadherins Drive Adhesion
- Chapter 20. Actin Cortex Mechanics and Cellular Morphogenesis
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Cell Press Reviews: Core Concepts in Cell Biology by in PDF and/or ePUB format, as well as other popular books in Biological Sciences & Biology. We have over 1.5 million books available in our catalogue for you to explore.