
- 816 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Liquid-Phase Extraction
About this book
Liquid Phase Extraction thoroughly presents both existing and new techniques in liquid phase extraction. It not only provides all information laboratory scientists need for choosing and utilizing suitable sample preparation procedures for any kind of sample, but also showcases the contemporary uses of sample preparation techniques in the most important industrial and academic project environments, including countercurrent chromatography, pressurized-liquid extraction, single-drop Microextraction, and more. Written by recognized experts in their respective fields, it serves as a one-stop reference for those who need to know which technique to choose for liquid phase extraction.
Used in conjunction with a similar release, Solid Phase Extraction, it allows users to master this crucial aspect of sample preparation.
- Defines the current state-of-the-art in extraction techniques and the methods and procedures for implementing them in laboratory practice
- Includes extensive referencing that facilitates the identification of key information
- Aimed at both entry-level scientists and those who want to explore new techniques and methods
Tools to learn more effectively

Saving Books

Keyword Search

Annotating Text

Listen to it instead
Information
Chapter 1
Milestones in the Development of Liquid-Phase Extraction Techniques
Colin F. Poole Department of Chemistry, Wayne State University, Detroit, MI, United States
Abstract
The evolution of apparatus and techniques for the liquid-phase extraction of gas, liquid, and solid samples is described and their virtues placed in a modern context of the requirement for streamlined, efficient, low-cost, and automated sample preparation methods. Methods for gas-liquid extraction include impingers, bubblers, and denuders. For liquid-liquid extraction the characteristic features of continuous liquid-liquid extraction, multistage countercurrent distribution, liquid-liquid chromatography, steam distillation-solvent extraction, solvent sublation, salting-out assisted liquid-liquid extraction, aqueous two-phase extraction, micelle-mediated extraction, liquid-membrane extraction, liquid-phase microextraction, and segmented continuous flow extraction are described. For solid-liquid extraction shake flask, Soxhlet, ultrasound-assisted, microwave-assisted, and pressurized-liquid extraction methods are described. Sample extraction and cleanup using liquid-liquid distribution is described for complex samples with an emphasis on multianalyte methods.
Keywords
Liquid-liquid partition; Solvent extraction; Selectivity; Distribution constants; Apparatus; Gas-liquid extraction; Solid-liquid extraction; Gas blowdown method; Evaporative concentrators; Multiresidue methods
1.1 Introduction
Liquid-phase or solvent extraction is a venerable technique at least as old as recorded history [1]. It is generally employed as a sample preparation technique in which target compounds are transferred from one phase, the sample or sample-containing phase, to a liquid phase where further processing and/or analysis occurs [2]. For solvent extraction the receiving phase is a liquid, and the sample is either a gas, liquid, or solid material, which is at least partially soluble in the liquid phase. Typical samples are composed of target compounds of interest, or analytes, with the remainder of the sample referred to as the matrix for which detailed information is not required. The general purpose of solvent extraction, therefore, is the selective isolation of the target compounds from the sample with minimal matrix contamination. Solvent extraction is often employed as an initial step in sample preparation and, if required, is followed by additional sample cleanup procedures, including further solvent extraction steps (liquid-liquid partition) or complementary separation techniques.
The selective extraction of target compounds by contact with a solvent is due to the relative solubility of target compounds in the solvent compared with the matrix. For a liquid or solid, this process is generally referred to as trituration or leaching and for a gas as stripping. The isolation of the target compounds from their matrix requires a two (or more)-phase system and a mechanism for phase separation. This implies an additional restriction of low mutual solubility for the sample (or sample phase) and the extraction solvent. For solids a mechanical separation in which the solvent is displaced from the region of the sample matrix by decantation, filtration, centrifugation, or forced flow is typically used. For gases a common arrangement is to disperse the sample as bubbles in the extraction solvent that then migrate to the surface of the liquid and collapse having transferred soluble or reactive compounds to the extraction solvent. For liquid samples the processing steps involve active contact; agitation or dispersion of the sample and extraction phases; settling or condensation to recreate the two (or more)-phase system by gravity, centrifugation, or other means; and finally mechanical separation of the phase enriched in the target compounds from the phase (or phases) containing mainly matrix. For manual extraction the earlier processing steps typically require only simple apparatus available in most laboratories, while more sophisticated, specialized, and less common equipment is required for automation [3–5].
Liquid-liquid distribution is a common technique accompanying solvent extraction in which a dissolved substance is transferred from one liquid phase to another immiscible (or partially immiscible) liquid phase in contact with it. The driving force for the transfer is the difference in the solubility of the target compounds in each phase of the biphasic system. For compounds that exist in the same chemical form in both phases and have attained equilibrium in the biphasic system, the ratio of the compound in both phases is described by the partition constant. This can be formally defined as the ratio of the activity of species A in the extract aA,1 to the activity in a second phase with which it is in equilibrium, aA,2
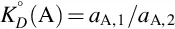
The value for KD° depends on the choice of standard states, temperature, and pressure. Distribution isotherms are generally linear over a reasonable concentration range. This allows concentrations (mol/L) at low to moderate concentrations to be substituted for activities in Eq. (1.1) for the calculation of KD°. Strictly speaking when concentrations are used in Eq. (1.1), the partition constant is referred to as the distribution constant KD [2], but this distinction is rarely made in the literature. For compounds that can exist in more than one chemical form in at least one phase, the distribution ratio, D, is used in place of the distribution constant. It is defined as the total concentration of a compound in the extraction phase to its total concentration in the other phase, regardless of its chemical form. It is the appropriate form of the distribution constant when secondary chemical equilibriums in one or both phases exert partial control over the distribution process [6–9]. Common examples of secondary chemical equilibriums encountered in liquid-liquid distribution are ionization, ion-pair formation, chelate formation, micelle formation, and aggregation. The distribution ratio depends on the distribution constant for each equilibrium process and is thus influenced by a wider range of experimental conditions than for a single partition mechanism. The distribution ratio is also used in connection with continuous flow processes operating at a steady state and does not imply that the system has achieved equilibrium.
The fraction of a compound extracted, E, in a single-stage batch process depends on both the distribution constant and the phase ratio, V. The latter is defined as the ratio of the volume of extraction solvent, VE, and sample solution, VS, contained in the extraction device
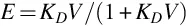
Extraction is favored by selecting conditions that result in a large value for KD and a suitable phase ratio. Large values of the phase ratio (VE ≫ VS) are favorable for the extraction of all compounds in a single-stage batch extraction but are rarely practical because the compounds are isolated in a too dilute solution. Typical experimental values for the phase ratio are closer to V = 1, and if KD is sufficiently large, V = 0.1–1.0. For compounds with a moderate distribution constant, a more efficient use of extraction solvent compared with a single batch extraction is provided by multiple extractions. This utilizes a number of sequential extractions of the sample with fresh extraction solvent, typically with a fixed phase ratio in which case the fraction extracted is given by
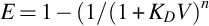
where n is the number of sequential extractions. When KDV = 10, 99% of the compound is extracted with n = 2; when KDV = 1, 99% of the compound is extracted with n = 7; and when KDV = 0.1, 50% of the compound is extracted with n = 7. Although large values of n favor exhaustive extraction, this approach is tedious, time-consuming, and labor-intensive for manual extraction. Automated batch methods, continuous flow methods, and countercurrent chromatography provide a more elegant option in this case [3, 4, 10]. Liquid-phase microextraction methods are characterized by an unfavorable phase ratio for exhaustive extraction (VS ≫ VE). In this case the extraction conditions typically correspond to negligible depletion of the target compound concentration, and the extracted amount is independent of the sample volume [11, 12]. Calibration is required to relate the concentration of extracted target compounds to the sample concentration.
In a typical batch extraction process, equilibrium is not...
Table of contents
- Cover image
- Title page
- Table of Contents
- Copyright
- Contributors
- Chapter 1: Milestones in the Development of Liquid-Phase Extraction Techniques
- Chapter 2: Solvent Selection for Liquid-Phase Extraction
- Chapter 3: Aqueous-Organic Biphasic Systems: Extraction of Organic Compounds
- Chapter 4: Fundamentals of Solvent Extraction of Metal Ions
- Chapter 5: Aqueous Two-Phase Systems
- Chapter 6: Octanol-Water Partition Constant
- Chapter 7: Surfactant-Based Extraction Systems
- Chapter 8: Microextraction With Supported Liquid Membranes
- Chapter 9: Totally Organic Biphasic Systems
- Chapter 10: Countercurrent Chromatography—When Liquid-Liquid Extraction Meets Chromatography
- Chapter 11: Soxhlet Extraction
- Chapter 12: Ultrasound and Microwave as Green Tools for Solid-Liquid Extraction
- Chapter 13: Pressurized Liquid Extraction
- Chapter 14: Quick, Easy, Cheap, Effective, Rugged, and Safe (QuEChERS) Extraction
- Chapter 15: Single-Drop Microextraction
- Chapter 16: Dispersive Liquid-Liquid Microextraction
- Chapter 17: Extraction With Ionic Liquids-Organic Compounds
- Chapter 18: Metal Ion Extraction With Ionic Liquids
- Chapter 19: Preanalytical Treatments: Extraction With Deep Eutectic Solvents
- Chapter 20: Environmental Applications
- Chapter 21: Application in Food Analysis
- Chapter 22: Extraction of Plant Materials
- Chapter 23: Biomedical Applications
- Chapter 24: Solvent Extraction for Nuclear Power
- Chapter 25: Continuous-Flow Extraction
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Liquid-Phase Extraction by Colin F. Poole in PDF and/or ePUB format, as well as other popular books in Physical Sciences & Analytic Chemistry. We have over one million books available in our catalogue for you to explore.