Breaking down large biomolecules into fragments in a controlled manner is key to modern biomolecular mass spectrometry. This book is a high-level introduction, as well as a reference work for experienced users, to ECD, ETD, EDD, NETD, UVPD, SID, and other advanced fragmentation methods. It provides a comprehensive overview of their history, mechanisms, instrumentation, and key applications. With contributions from leading experts, this book will act as an authoritative guide to these methods.
Aimed at postgraduate and professional researchers, mainly in academia, but also in industry, it can be used as supplementary reading for advanced students on mass spectrometry or analytical (bio)chemistry courses.

eBook - ePub
Advanced Fragmentation Methods in Biomolecular Mass Spectrometry
Probing Primary and Higher Order Structure with Electrons, Photons and Surfaces
- 342 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Advanced Fragmentation Methods in Biomolecular Mass Spectrometry
Probing Primary and Higher Order Structure with Electrons, Photons and Surfaces
About this book
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
Physical SciencesSubtopic
BiologyChapter 1
Modern Mass Spectrometry and Advanced Fragmentation Methods
a Department of Chemistry, Institute of Chemistry and Biochemistry, Technical University of Darmstadt64287 Darmstadt, Germany
b Mass Spectrometry Laboratory, MolSys Research Unit, University of Liège4000 LiègeBelgium
c School of Engineering, University of WarwickCoventry CV4 7ALUK
[email protected]
[email protected]
1.1 Introduction
In 2019, it had been exactly 150 years since Dmitri Mendeleev compiled the first version of the periodic table of elements. He did this by arranging the then-known elements (around 60) by increasing atomic mass and noting the recurrence of certain chemical properties. Today, the number of identified elements – including synthetic ones – has roughly doubled, and the periodic table is perhaps the most recognisable achievement of all of chemistry to a general audience. For many of us, it was the first thing we saw the very first time we set foot in a chemistry classroom. Because of how universally important the periodic table is, any 18-year-old with even a limited interest in science is likely to know that every element in this table is associated with a number that indicates its atomic mass, and that this can be easily used to calculate the molecular mass of chemical compounds. They might even know that most elements occur in different variants, called isotopes, that have slightly different masses, and that the unit for atomic and molecular mass, the Dalton (Da), is one-twelfth of the mass of a carbon-12 atom.
If our hypothetical young person is interested enough to pursue a scientific study in higher education, they will likely learn that it is relatively fast and straightforward today to determine the molecular mass of a compound using a specialised instrument called a mass spectrometer. For most users, mass spectrometry (MS) is merely a tool used to confirm the nominal molecular mass of a compound. While the technique can be used in this way, this only scratches the surface of its capabilities. By high-resolution, high-accuracy mass measurement, it is often possible to determine a unique elemental composition for ions with molecular mass up to a few hundred Da. At sufficiently high resolution, exact isotopic variants can be distinguished, i.e., it can be determined to which nucleus a neutron was added. This is due to the different binding energies for different nuclei, which can be measured by mass spectrometry due to Einstein's famous equation, E=mc2. These differences are usually of the order of one to a few mDa. Going to even higher resolution, it becomes possible to elucidate reactions by monitoring the movement of individual electrons. This might be surprising, given that the mass of an electron is only 0.00055 Da; however, measurement of ions of up to several hundred Da with this level of accuracy is routine today using high-performance instruments. At some point in the future, it might even become possible to measure chemical binding energies directly from accurate mass measurement using the same principle as for nuclear binding energies. As one electronvolt corresponds to a mass equivalent of ca. 1.1 nDa, this is beyond the capabilities of even the best instruments in use today.
Of course, while a significant amount of valuable information can be obtained from accurate measurement of the mass of an intact ion alone, the real strength of modern biomolecular MS lies in its ability to isolate an ion in the gas phase, break one or more of the chemical bonds present in the ion, and then measure the mass of the fragments. For both precursor and fragment masses, the above-mentioned benefits of accurate mass determination apply. A number of excellent introductory texts to mass spectrometry exist1,2 and a familiarity with basic concepts is assumed in this book. Some key concepts such as commonly used ionisation methods and mass analysers in modern biomolecular MS, and figures of merit in MS will be briefly recapitulated here.
1.2 Ionisation Methods
A mass spectrometer essentially consists of the following components: (1) an ion source, (2) a mass analyser, (3) a detector, and (4) a data acquisition system. Fundamentally, all mass spectrometers measure the mass-to-charge, or m/z value of gas-phase ions. Generally, this is because the interaction of the ions with an electromagnetic field accelerates them, while their inertia (i.e., mass) causes them to resist this acceleration. The magnitude of acceleration is proportional to charge, and inversely proportional to mass, and this is why, even though a variety of mass analysers have been developed relying on a range of physical principles, they all provide a measure of m/z. This of course assumes that the ions have a sufficiently large mean free path that their motion is not meaningfully affected by collisions with background gas molecules during mass analysis. For this reason, measurements are performed in a vacuum, and the vacuum system is usually either listed as a separate component, or understood to be part of the mass analyser.
Producing gas-phase ions is not trivial, and a range of methods has been developed over the years. Electron (impact) ionisation was developed in the early 20th century, first for solids,3 and later for gaseous4 samples. Unfortunately, this method is not appropriate for polar, high molecular mass analytes such as most biomolecules. Today, solid samples are more often analysed using matrix-assisted laser desorption/ionisation (MALDI),5,6 secondary ion mass spectrometry (SIMS),7 or laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS).8 For biomolecular MS, samples are most commonly introduced in solution, and the most effective ionisation method in this case is (nano-) electrospray ionisation (ESI) (see Figure 1.1).9,10 In this method, a potential difference is applied between a capillary containing the sample solution (at atmospheric pressure) and the inlet of the mass spectrometer. The resulting electrostatic force leads to deformation of the liquid surface to form a Taylor cone, which, when the attraction becomes sufficiently large to overcome the surface tension of the liquid, releases small droplets with an excess of charge. The exact mechanism for what happens next is still debated,11–13 but this process ultimately results in formation of desolvated, multiply protonated (in positive ion mode) or deprotonated (negative mode) ions in the gas phase.
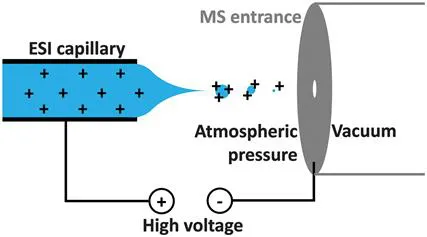
Figure 1.1 Principle of electrospray ionisation for cation formation.
1.3 Figures of Merit for Mass Spectrometry
The two most commonly cited figures of merit in mass spectrometry are mass accuracy and resolution. Mass accuracy is simply the difference between the expected and measured m/z value. This is typically expressed as a fraction of the expected value rather than an absolute number, and as it tends to be a very small deviation using modern instrumentation, it is usually expressed as parts-per-million (ppm). It is important to emphasise at this point that this use of ‘ppm’ refers to accuracy of measured m/z values, and is unrelated to concentration or sensitivity. For example, the exact mass of singly protonated arginine (with all elements in their lightest isotopic variant) is 175.118952 Da. If our measurement is one ppm (or 0.0001%) too high, we will instead obtain a value of 175.119127, i.e., a deviation of less than 0.2 mDa, or 30% of the mass of an electron. This level of accuracy is typical for commercial Fourier transform ion cyclotron resonance (FTICR), orbitrap, and time-of-flight (TOF) instruments which are able to record data accurate to a few ppm (vide infra). The general formula is:
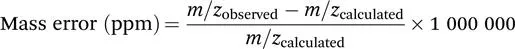
As the calculated mass is subtracted from the observed value, a positive ppm error is associated with overestimating the mass, and a negative value with underestimating it.
Resolution is also referred to as resolving power, and some authors interpret these two terms as having a subtly different meaning. In the context of mass spectrometry, these terms refer to peak width. Except in very specific single-ion applications, a signal in MS is always generated by many ions. Due to inherent instrumental factors including field inaccuracies and signal measurement errors, ions with identical m/z will be observed at slightly different m/z values, resulting in a signal in the spectrum possessing non-zero peak width. It should be noted that some commercial instruments actually do display zero-width bars (known as a ‘centroid spectrum...
Table of contents
- Cover
- Title
- Copyright
- Preface
- Contents
- Chapter 1 Modern Mass Spectrometry and Advanced Fragmentation Methods
- Chapter 2 Mechanism and Implementation of Electron Capture and Electron Transfer Dissociation
- Chapter 3 ECD/ETD for Sequencing of Peptides and Denatured Proteins
- Chapter 4 Structural Studies using Electron-based Fragmentation Methods and Chemical Labelling of Proteins
- Chapter 5 Investigating 3D Structures of Native Proteins and Complexes through Electron-based Dissociation
- Chapter 6 Electron Detachment Dissociation (EDD) and Negative Electron Transfer Dissociation (NETD)
- Chapter 7 Electron Photodetachment and the Radical Fragmentation Route for Negative Ions
- Chapter 8 Elucidating Biomolecular Structure Through Bond-selective Radical Fragmentation
- Chapter 9 Structural Analysis of Lipids Using Advanced Tandem MS Methods
- Chapter 10 Photoactivation and Dissociation in Biomolecular Mass Spectrometry
- Chapter 11 Surface-induced Dissociation in Biomolecular Mass Spectrometry
- Subject Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Advanced Fragmentation Methods in Biomolecular Mass Spectrometry by Frederik Lermyte in PDF and/or ePUB format, as well as other popular books in Physical Sciences & Biology. We have over 1.5 million books available in our catalogue for you to explore.