Over the past decade, the understanding of the processes involved in the regulation of gonadotropin-releasing hormone and its dysfunction has greatly increased. As new regulatory peptides have been identified, the underlying causes of central hypogonadism have multiplied, and the area has become increasingly complex. The reversibility of even genetically determined hypogonadotropic hypogonadism has become more firmly established, and clinical studies have greatly expanded our understanding of basic physiological pathways. Structuring this mass of new knowledge in thirteen comprehensive chapters, a group of renowned experts, representing the principal international research groups, take stock of the most recent progress.This up-to-date overview helps scientists and clinicians to plan future research and treat patients with delayed puberty, hypogonadotropic hypogonadism and other forms of central reproductive disorders.

eBook - ePub
Kallmann Syndrome and Hypogonadotropic Hypogonadism
- 184 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Kallmann Syndrome and Hypogonadotropic Hypogonadism
About this book
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
MedicineSubtopic
Genetics in MedicineQuinton R (ed): Kallmann Syndrome and Hypogonadotropic Hypogonadism.
Front Horm Res. Basel, Karger, 2010, vol 39, pp 78–93
Front Horm Res. Basel, Karger, 2010, vol 39, pp 78–93
______________________
Biological Actions and Interactions of Anosmin-1
Catherine Choy · Soo-Hyun Kim
Division of Basic Medical Sciences, St. George’s Medical School, University of London, London, UK
______________________
Abstract
Kallmann syndrome is a multigenic human developmental disorder where the molecular pathogenesis is still only partially understood and there is no single unifying animal model. The protein anosmin-1, encoded by the KAL1 gene, is associated with the X-linked form of the disease. The biology and molecular structure of anosmin-1 has been investigated systematically over the years by various cell culture experiments, biochemical analyses and animal models. Anosmin-1 is an extracellular matrix-associated protein which plays pleiotropic roles in neuronal development, migration and organogenesis.
Copyright © 2010 S. Karger AG, Basel
When idiopathic hypogonadotropic hypogonadism (IHH) is combined with anosmia or hypoanosmia, it is defined as Kallmann syndrome (KS). KS is a relatively rare congenital heterogeneous disorder, affecting approximately 1 in 8,000 males [1] and with a 3- to 5-fold excess of male cases over female reported by most investigators. The association of hypothalamic gonadotropin-releasing hormone (GnRH) deficiency and olfactory bulb (OB) dysgenesis is presumably caused by the disruption of the olfactory GnRH neuronal migration during early embryogenesis [2]. KS can be transmitted monogenically in either X-linked, autosomal dominant or autosomal recessive modes of inheritance, but the majority of cases are sporadic. However, recent studies have highlighted the more complex nature of the genetics of this disease [3, 4]. So far, six different factors have been identified as the underlying genetic defects: anosmin-1 (KAL1) [5, 6], fibroblast growth factor receptor 1 (FGFR1) [7] anditsligand (FGF8) [10], prokineticin 2 (PROK2) and its receptor (PROKR2) [8], and chromodomain helicase DNA binding protein 7 (CHD7) [9]. However, the genetic basis of the majority of cases probably still remains unaccounted for. KS is thus an oligogenic trait involving a number of key signaling pathways, with further genetic defects remaining to be discovered.
KAL1 was first identified as the genetic locus for X-linked KS nearly two decades ago [5, 6]. During this period, the exact function and regulation of the KAL1 gene and its encoded protein named anosmin-1 have been the focus of much research. The KAL1 gene localised to Xp22.3, contains 14 exons encompassing 220 kb of genomic DNA and 2,043 bases of coding sequences. KAL1 orthologs have been identified or predicted in many vertebrates and invertebrates including fruit fly, worm, chicken, dog, fish, frog, monkey and chimpanzee [11]. Despite full sequencing of the mouse genome, an ortholog remains unidentified, potentially due to such significant sequence divergence from human that the value of any mouse model would anyway be severely limited. However, studies using Caenorhabditis elegans, zebrafish, medaka and Drosophila melanogaster have provided substantial new information. Furthermore, the sequence alignment of known KAL1 genes shows high levels of homology involving specific protein domains, suggesting important functions of these regions (fig. 1). This review will focus on several recent studies providing significant new insights into the functions of anosmin-1, in particular its molecular structure, biological activities and interacting partners that may assist or enable its function. Other KS-associated molecules and the complex nature of the phenotype-genotype relations in KS will be reviewed in separate chapters of this volume.
Molecular Structure of the Functional Domains of Anosmin-1
The open reading frame of human KAL1 cDNA encodes a 680-amino acid protein with a conceptual molecular weight of 74 kDa, which subsequently undergoes N-glycosylation contributing to an observed molecular weight of 85-100 kDa of the mature protein [12–14]. There has been no direct confirmation regarding which of the putative glycosylation sites are involved in this post-translational modification. However, it has been shown that glycosylation has an eminent role in functional protein folding and secretion, as well as other protein binding interactions in cellular signalling events [15]. Intracellular localisation studies have demonstrated that anosmin-1 is an extracellular matrix (ECM)-associated secretory protein, showing accumulation along the plasma membrane and within endoplasmic vesicles, as well as in the surrounding medium scattering to adjacent cells [16, 17]. These observations are consistent with the lack of any hydrophobic transmembrane domains or glycosyl phosphotidyl inositol anchorage site in the structure of anosmin-1. Instead, it comprises a signal peptide, a cysteine-rich N-terminal region, a whey acid protein (WAP)-like four-disulphide core motif, followed by four tandem fibronectin type III (FnIII)-like domains and a histidine-rich C-terminus (fig. 1).
The WAP domain in anosmin-1 contains four-disulphide core motif, similar to those found in secretory leukocyte protease inhibitor, elafin, human secretory protein 4 (HE4), Westmead DMBA8 nonmetastatic cDNA 1 (WDNM1) and 20-kDa prostate stromal protein (ps20). Secretory leukocyte protease inhibitor and elafin are anti-serine protease and anti-microbial molecules with a role in innate immune defence, wound healing and tissue remodelling, and elafin degrades ECM proteins such as elastin [18]. HE4 is a serological tumour biomarker used to predict ovarian cancer and has a presumptive role in natural immunity [19]. WDNM1 is an extracellular proteinase inhibitor whose downregulation has been linked with metastatic rat mammary adenocarcinomas [20]. Finally, ps20 has protease-inhibitory activity and can stimulate endothelial cell migration and promotes angiogenesis and tumour growth in a prostate cancer model [21], though in a different study, ps20 was shown to inhibit the proliferation of prostate carcinoma cells [22]. In all, WAP family of proteins exhibit protease inhibitory activity, regulate cell proliferation, differentiation and tissue re-modelling in various tissue types. That the WAP domain is evolutionarily conserved in all anosmin-1 sequences suggests that anosmin-1 might also be capable of some of these functions. However, it has been shown that anosmin-1 promoted, rather than inhibited, the proteolytic activities of urokinase type plasminogen activator (uPA) in vitro, while exogenous addition of anosmin-1 induced cell proliferation in prostate carcinoma cell line PC3 [13]. The activity of the WAP domain might be affected in the C172R or C163Y missense mutations found in KS patients, since these substitutions are thought to disrupt the conserved C151-C163, C157-C172disulphide bonds, probably affecting the protein folding or structural stability of this region.
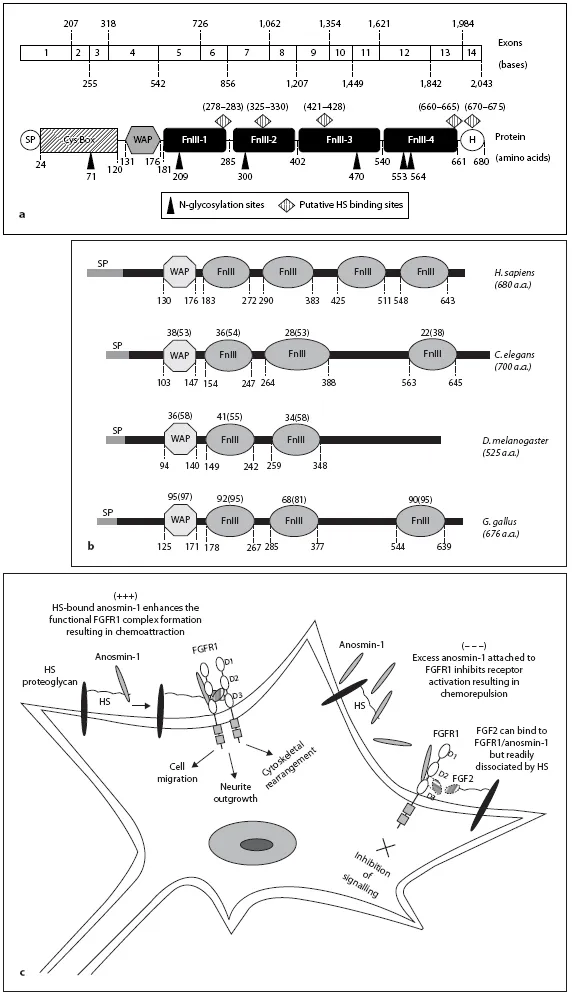
Fig. 1. a Structure of KAL1 gene and anosmin-1 protein. Schematic representation of human KAL1 cDNA and anosmin-1 protein. The KAL1 gene is composed of 14 exons encoding a 2.4 kb of transcript and 680 amino acids. The six putative N-glycosylation sites and five potential HS binding sites are indicated and amino acid residues for each are shown. Nucleotide numbers for each exon are also shown. There is a signal peptide (SP) followed by a cysteine rich N-terminus (Cys Box),WAP-like four-disulphide core motif and four FnIII domains ending with a histidine (H)-rich C-terminus. The WAP domain and the first FnIII domain are functionally essential. Anosmin-1 lacks a transmembrane anchorage, thus is likely to be ECM-associated and secreted. b Conservation of functional domains of anosmin-1 through evolution. The domain structures of the anosmin-1 proteins based on SMART (http://smart.embl-heidelberg.de/smart). Homo sapiens (sequence ID:P23352), C. elegans (sequence ID: Q8WS94), D. melanogaster (sequence ID: Q9VCC7) and Gallus gallus (sequence ID: UPI000012DC04) are aligned. The relative positions of each domain in amino acid residues are shown as the numbers below the dotted lines. The numbers above each domain represent the percentage of identities and positives (shown in parentheses) compared to human, which is determined by alignment of the corresponding protein sequences of each domain using BLAST (www.ncbi.nlm.nih.gov/blast). c Putative mechanisms of molecular interaction between anosmin-1, FGFR1, FGF2 and HS on the cell surface. When HS-bound anosmin-1 associates with an otherwise transient complex between FGFR1 and FGF2, a stable anosmin-1/FGFR1/FGF2/HS complex forms leading to activation of the receptor and the consequent network of FGFR1 signalling pathways as indicated. On the other hand, when there is high-level expression of anosmin-1 potentially exceeding the binding capacity of the local HS, free anosmin-1 attaches with high affinity to FGFR1, preventing the formation of an FGF2/ FGFR1/HS signalling complex.
D. melanogaster has two, chicken and C. elegans have three and human has four FnIII domains in anosmin-1 protein (fig. 1b). The FnIII domains of anosmin-1 have significant homology with members of the cell adhesion molecule (CAM) family including fibronectin, neural CAM (NCAM), L1 and tenasin. These molecules are usually associated with extracellular and cell-cell interaction, adhesion, proliferation and migration during neuronal development, suggesting a similar involvement of anosmin-1 in these cellular processes. FnIII is a small autonomous folding unit which occurs in many proteins involved in ligand binding and dimerisation [23], and it has been shown that FnIII domains in NCAM, L1, N-Cadherin, Sef and Xenopus fibronectin leucine-rich transmembrane protein 3 (XFLRT3) play essential roles in their binding interactions with FGFR1 [24–28]. Consistently, our recent study demonstrated that anosmin-1 directly binds to FGFR1 through the FnIII domain [17]. Since the arrangement of the six domains of anosmin-1 in solution state appears extended, with flexible inter-domain linkers, it has been speculated that anosmin-1 may interact with multiple molecules providing a binding platform or scaffolding for potential multiprotein complex formation on the cell surface [29]. There are five putative heparan sulphate (HS) binding sites amongst or within the individual FnIII domains of anosmin-1 (fig. 1a). HS-mediated cell surface association is essential for anosmin-1’s activity [30, 31]. HS acts as a receptor or co-receptor to regulate the biological activities of HS-binding proteins to promote dimerisation of its ligand and receptor, induce receptor phosphorylation and stabilise an active ligand-receptor complex formation. Interaction studies between the HS and FGF system is one of the best studied examples [32] and will be discussed further in relation to anosmin-1 in the later section. The predominant loss-of-function mutations (frameshift, deletions) with 10 missense mutations are identified in the WAP and the first FnIII domains so far and five missense mutations identified have been mapped closely to the putative HS binding sites. It is speculated that N267K (in first FnIII domain) and E514K (in third FnIII domain) mutations associated with X-KS cases may enhance HS binding as these are neutral/negative to positive amino acid substitutions and the increased HS binding may inhibit the molecular movement or flexibility of anosmin-1 in the ECM. An F517L substitution (in third FnIII domain), although occurs...
Table of contents
- Cover Page
- Front Matter
- Molecular Characterization and Phenotypic Expression of Mutations in Genes for Gonadotropins and Their Receptors in Humans
- Role of Kisspeptin/GPR54 System in Human Reproductive Axis
- Biology of Kisspeptins
- Role of Fibroblast Growth Factor Signaling in Gonadotropin-Releasing Hormone Neuronal System Development
- FGFR1 Mutations in Kallmann Syndrome
- Biology of KAL1 and Its Orthologs: Implications for X-Linked Kallmann Syndrome and the Search for Novel Candidate Genes
- Biological Actions and Interactions of Anosmin-1
- Genotype and Phenotype of Patients with Gonadotropin-Releasing Hormone Receptor Mutations
- Hypogonadotropic Hypogonadism and GNRH1 Mutations in Mice and Humans
- Kallmann Syndrome Caused by Mutations in the PROK2 and PROKR2 Genes: Pathophysiology and Genotype-Phenotype Correlations
- Neurokinin B and Its Receptor in Hypogonadotropic Hypogonadism
- Complex Genetics in Idiopathic Hypogonadotropic Hypogonadism
- Rarer Syndromes Characterized by Hypogonadotropic Hypogonadism
- Concluding Remarks
- Author Index
- Subject Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Kallmann Syndrome and Hypogonadotropic Hypogonadism by R. Quinton, Federica Guaraldi,Giovanni Corona in PDF and/or ePUB format, as well as other popular books in Medicine & Genetics in Medicine. We have over 1.5 million books available in our catalogue for you to explore.