Before Initiation
A risk-benefit analysis for a clinical trial is provisionally based on the preclinical phase of the medicinal product. The sponsor-investigator team needs to evaluate the toxicological tests and results as well as submit the data to the competent health authorities, with a projection of all the possible risks for the proposed trial subjects.
This provisional projection, however, cannot provide a reliable statement to the ‘true’ risk-benefit assessment of an investigational medicinal product (IMP).
Regulatory Reference
! International Conference on Harmonization and Good Clinical Practice (ICH GCP) Guideline, Chapters 2.2, 3.1.4, 3.3.8 (b), 4.8.10 (i), 4.10.2, 5.19.3 (b), 6.2.3, 7.1, 7.3.6 (b)
! Declaration of Helsinki
! EU Directive 2001/20/EC, Art. 1 (2), (18), Art. 4, Art. 5
Responsibilities
The sponsor-investigator team must collect all available information to be able to undertake an adequate risk-benefit analysis. Usually, the Investigator’s Brochure (IB) and/or the Summary of Product Characteristics (SmPC) - the latter available when the medicinal product planned to be further investigated is already on the market - are the main documents for which the sponsor-investigator should request the medicinal product manufacturer to provide.
More often is the case when the sponsor-investigator has no prior clinical data to accomplish a true risk-benefit analysis. Thus, this analysis is an evaluation of the available knowledge. The important clinical data will be gathered during the trial.
There may be other available information sources: sponsor-investigators should check the supplemental information in the SmPC and ask the manufacturer of the medicinal product if there are any prior risk-benefit analyses.
see 11
Investigator’s Brochure/Summary of Product Characteristics
During Study
One of the most important concepts in a clinical trial is to ensure and protect the well-being of the involved trial subjects. Therefore, at all times, you must act to minimize the risk to human beings, regardless of the trial performance.
During the entire study, the data needs to be reviewed and evaluated routinely. Usually, a team of experts (or a formal Data Monitoring Committee [DMC]) review the data and decide on a periodic basis, whether the study should continue as planned.
Regulatory Reference
! ICH GCP Guideline, Chapters 2.2, 3.1.4, 3.3.8 (b), 4.8.10 (i), 4.10.2, 5.19.3 (b), 6.2.3, 7.1, 7.3.6 (b)
! Declaration of Helsinki
! EU Directive 2001/20/EC, Art. 1 (2), (18), Art. 4, Art. 5
Responsibilities
Definition
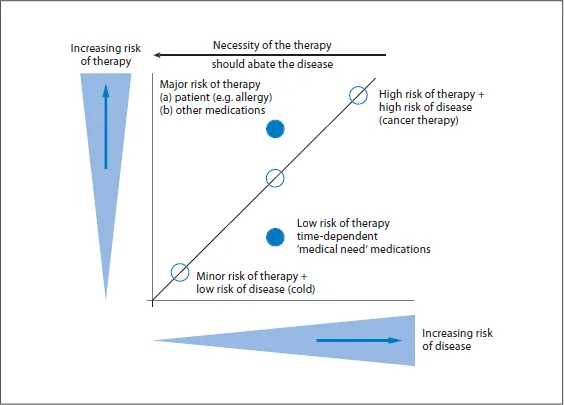
The risk-benefit-analysis describes, on the one hand, the proportion of the potential treatment efficacy, and on the other hand, the proportion of all the possible types of risks to the study patient - whether due to the quality, safety or efficacy of the investigational product. This proportion is crucial for the approval.
Importance
To obtain a meaningful risk-benefit analysis, there are two important considerations. Not only is the most favourable benefit of the investigational product for the trial subject of importance, but also the cognition of potential risks of therapy.
There are some diseases (e.g. cancer, cardiac infarction) that have a very high mortality. In this instance, it might be beneficial to take a higher risk with the therapy. For diseases that are symptomatic but do not jeopardize the overall health of the patient (e.g. common cold), it has to be ensured that the risk is considerably less than the benefit. In the figure, the ideal ratio would be in the lower right-hand corner, i.e. the benefits of treatment outweigh the risks.
Deliberations in Risk-Benefit Analysis of Pharmacotherapy.
The ICH GCP assumes that the risk-benefit ratio is of prime importance for each trial subject as well as for the public health. However, an official criterion for the ratio calculation does not exist. It always is an individual decision how to make this analysis or calculation. At the time of the approval, it is sufficient that the evaluation of this ratio is provisional.
Responsibilities in Each Study Phase
a Phase I:
The sponsor-investigator, the regulatory health authorities and an Independent Ethics Committee (IEC) are competent to estimate the ratio between risk and benefit by reference to the data recording safety and effectiveness.
b Phase II/III:
The sponsor-investigator has to inform the health authority and the IEC about sudden unexpected serious adverse reactions (SUSARs). Furthermore, the sponsor-investigator must supply a safety report annually. Based on those documents, the health authority and the IEC have to decide if the risk-benefit ratio is acceptable to continue the trial.
see 19
Pharmacovigilance
Advice - Hints and Tips
• The risks and the benefits have to be reviewed and discussed by the sponsor-investigator team. The data included in the review should be balanced for the planned trial. Ideally, this analysis should take place prior to starting to write the protocol and agreed to by the team.
• Nevertheless, the risk-benefit ratio has to be monitored carefully throughout the clinical trial (if possible, by a pharmacovigilance expert); if the ratio turns negative, you have to consider the consequences for the trial continuation.
• There has to be a conclusive regulatory consideration from the health authorities regarding the risk-benefit ratio. Only after a positive decision, the investigational IMP can be launched and made available to the pharmaceutical market.
see 2
Study Types and Study Design
see 19
Pharmacovigilance
Before Initiation
The scientific quality and validity of a clinical trial is primarily determined by the study design. The design must be planned very carefully, because it is difficult to correct inconsistencies afterward. The academic details of planning is not the only determinant of quality - the financial, organizational, logistical and personnel elements of the clinical trial must also be considered in advance by the sponsor-investigator. Early and painstaking considerations in the study design can also prevent influences (so-called bias) from distorting the results of the pre-planned statistical test procedures.
Regulatory Reference
! ICH GCP Guideline, sections 4.2.1 and 2, 4.7
! EU Directive 2001/20/EC, Art. 2 a, b, c
Responsibilities
Study Types
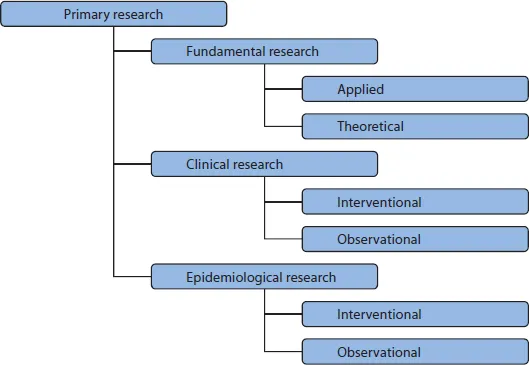
In principle, two categories of studies exist: the science differentiates between studies of primary and secondary data. This book does not deepen the topic of secondary data studies, because the research with secondary data only looks at studies that have been completed. The research with primary data means the initiation of a trial with recording of primary data. There are also other differences:
The investigator-initiated trial is a clinical trial involving human subjects; it is always a part of interventional clinical research.
Study Population
To make the right conclusions with regards to the general population using statistical test procedures, the sponsor-investigator has to recruit a representative study population. The principle behind this is that the initial starting position or baseline for the clinical trial is, in essence, the definition of the target group for treatment.
Study Design
The primary aim of a study design is an outcome with a high explanatory power. The gold standard of clinical research is the randomized controlled trial or RCT.
The first step is the randomization of the treatment groups, because the experimental units are never identical. By using a random mechanism, the subjects are allocated to the treatment gro...