The vast majority of drugs are organic molecular entities. A clear understanding of the organic chemistry of drug degradation is essential to maintaining the stability, efficacy, and safety of a drug product throughout its shelf-life. During analytical method development, stability testing, and pharmaceutical manufacturing troubleshooting activities, one of the frequently occurring and usually challenging events would be the identification of drug degradants and understanding of drug degradation mechanisms and pathways. This book is written by a veteran of the pharmaceutical industry who has first-hand experience in drug design and development, drug degradation mechanism studies, analytical development, and manufacturing process troubleshooting and improvement. The author discusses various degradation pathways with an emphasis on the mechanisms of the underlying organic chemistry, which should aid greatly in the efforts of degradant identification, formulation development, analytical development, and manufacturing process improvement. Organic reactions that are significant in drug degradation will first be reviewed and then illustrated by examples of drug degradation reported in the literature. The author brings the book to a close with a final chapter dedicated to the strategy for rapid elucidation of drug degradants with regard to the current regulatory requirements and guidelines. One chapter that should be given special attention is Chapter 3, Oxidative Degradation. Oxidative degradation is one of the most common degradation pathways but perhaps the most complex one. This chapter employs more than sixty drug degradation case studies with in-depth discussion in regard to their unique degradation pathways. With the increasing regulatory requirements on the quality and safety of pharmaceutical products, in particular with regard to drug impurities and degradants, the book will be an invaluable resource for pharmaceutical and analytical scientists who engage in formulation development, analytical development, stability studies, degradant identification, and support of manufacturing process improvement. In addition, it will also be helpful to scientists engaged in drug discovery and development as well as in drug metabolism studies.

- 306 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Organic Chemistry of Drug Degradation
About this book
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
MedicinaSubtopic
FarmacologiaCHAPTER 1
Introduction
1.1 Drug Impurities, Degradants and the Importance of Understanding Drug Degradation Chemistry
A drug impurity is anything that is not the drug substance (or active pharmaceutical ingredient, API) or an excipient according to the definition by the US Food and Drug Administration (FDA).1 Impurities can be categorized into process impurities, drug degradation products (degradants or degradates), and excipient and packaging-related impurities. Process impurities are produced during the manufacture of the drug substance and drug product, while degradants are formed by chemical degradation during the storage of the drug substances or drug products. The storage conditions are typically represented by the International Conference on Harmonisation (ICH)- and World Health Organization (WHO)-recommended stability conditions which simulate different climatic zones of the world.2,3 Certain process impurities can also be degradants, if they continue to form in storage under stability conditions. Packaging-related impurities, also called leachables, are typically various plasticizers, antioxidants, UV curators, and residual monomers that leach out of the plastic or rubber components and labels of the package/container of a drug product over time.
Those process impurities that are not degradants may be controlled or eliminated by modifying or changing the process chemistry. On the other hand, control or minimization of drug degradants requires a clear understanding of the drug degradation chemistry, which is not only critically important for developing a drug candidate but also for maintaining the quality, safety, and efficacy of an approved drug product. Specifically, knowledge of drug degradation is not only vital for developing adequate dosage forms that display favorable stability behavior over the registered product shelf-life, but is also critical in assessing which impurities would be most likely to be significant or meaningful degradants so that they can be included in the specificity mixture when developing and validating stability-indicating analytical methodologies. A common problem in the development of stability-indicating HPLC methods using stress studies (or forced degradation) is a lack of proper evaluation if the stress-generated degradants would be real degradants or not. From a practical point of view, the real degradants are those that can form under long term storage conditions such as the International Conference on Harmonisation (ICH) stability conditions.2 On the other hand, various artificial degradants can be generated during stress studies, in particular when excessive degradation is rendered or the stress conditions are not consistent with the degradation pathways of the drug molecule under the usual stability conditions. For example, forced degradation of a ketone-containing drug, pentoxifylline, using 30% hydrogen peroxide at room temperature for eight days produced a geminal dihydroperoxide degradation product (Scheme 1.1).4 This compound is highly unlikely to be a real degradant of the drug product.
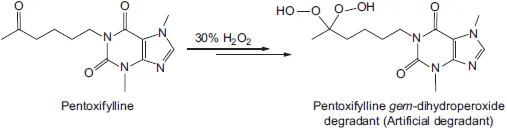
Scheme 1.1
This book is devoted to increasing our understanding and knowledge of the organic chemistry of drug degradation. The knowledge derived from this endeavor should also be beneficial for the elucidation of drug metabolite structures and bioactivation mechanisms. Most drugs undergo at least certain level of metabolism,5 that is, chemical transformation catalyzed by various enzymes. Except in the case of pro-drugs, drug metabolites can be considered as drug degradants formed in vivo. Chemical degradation and drug metabolism can produce the same degradants, even though they may go through different reaction intermediates or mechanisms. In vitro chemical reactions have been used to mimic enzyme-catalyzed drug metabolism processes, in order to help elucidate the enzymatic mechanisms for the catalysis.6 On the other hand, understanding the mechanisms of drug metabolism may also facilitate the elucidation of drug degradation pathways in vitro.
Regardless of their origins, certain drug degradants can be toxic, which is one of the main contributors to undesirable side effects or adverse drug reactions (ADR) of drugs.7 In the early stage of drug development, the degradants (including metabolites) and degradation pathways (or bioactivation pathways in the case of reactive metabolites) of a drug candidate need to be elucidated, followed by toxicological evaluation of these degradants. Dependent upon the outcome of the evaluation, the structure of the drug candidate may have to be modified to avoid the formation of a particular toxicophore based on the understanding of the degradation chemistry (or bioactivation pathways) elucidated. Failure to uncover toxic degradants, usually the low level ones, in the early development stage can lead to hugely costly failure in later stage clinical studies or even withdrawal of an approved drug product from the market.
1.2 Characteristics of Drug Degradation Chemistry and the Scope of this Book
The vast majority of therapeutic drugs are either organic compounds or biological entities. The latter drugs include protein and nucleic acid (RNA and DNA)-based drugs which are biopolymers comprising small molecule building blocks. This book focuses on the organic chemistry aspect of drug degradation, in particular, the mechanisms and pathways of the chemical degradation of both small and large molecule drugs under real life degradation scenarios, as represented by the usual long term stability conditions. Stress studies or forced degradation can help elucidate the structures of real degradants and the degradation pathways of drugs. Nevertheless, caution needs to be taken in differentiating the real and artificial degradants. This subject will be discussed in detail in Chapter 8, Strategies for Elucidation of Degradant Structures and Degradation Pathways.
Drug degradation chemistry differs from typical organic chemistry in several ways. First, the yield of a drug degradation reaction is usually very low, from approximately 0.05% to a few percentage points at the most. Dependent upon the potencies and maximum daily dosages of the drugs, ICH guidelines require that the impurities and/or degradants of a drug be structurally elucidated, once they exceed certain thresholds, which are typically between 0.05% and 0.5%, relative to the drug substances.8,9 For potential genotoxic impurities, they need to be characterized and controlled at a daily maximum amount of 1.5μg for drugs intended for long term usage.10 Such low yields would be meaningless from the perspective of the regular organic chemistry. Second, due to the low yields and limited availability of samples, particularly stability samples of formulated drugs, the quantity of a drug degradant is usually extremely low, posing a serious challenge for its isolation and/or characterization. Despite the advent of sensitive and powerful analytical methodologies such as high resolution tandem liquid chromatography-mass spectrometry (LC-MS/MS), liquid chromatography-nuclear magnetic resonance (LC-NMR), and cryogenic micro NMR probes, the identification of drug degradants remains one of the most challenging activities in pharmaceutical development.11 Third, the typical conditions and “reagents” of drug degradation reactions are limited in scope. For example, the ICH long term stability conditions for different climatic zones specify the requirements for heat and moisture (relative humidity, RH), for example, 25°C/60% RH and 30°C/65% RH, while the ICH accelerated stability condition requires heating at 40°C under 75% RH. In addition to moisture, the other most important “reagent” in drug degradation reactions is molecular oxygen. Since molecular oxygen is ubiquitous and difficult to remove from drug products, oxidative degradation of drugs is one of the most common degradation pathways. Often, the impact of molecular oxygen can be indirect. For example, a number of polymeric drug excipients such as polyethylene glycol (PEG), polysorbate, and povidone, are readily susceptible to autooxidation, resulting in the formation of various peroxides including hydrogen peroxide.12–14 These peroxides can cause significant drug degradation once formulated with drug substances containing oxidizable moieties. In contrast, reductive degradation is rarely seen in drug degradation reactions owing to the lack of a reducing agent in common drug excipients that is strong enough to cause meaningful reductive degradation. Other possible “reagents” in drug degradation reactions are usually limited to drug excipients and their impurities. For example, excipients consisting of oligosaccharides and polysaccharides with reducing ends, such as lactose and starch, are frequently used in drug formulation. The aldehyde functionality of these excipients can react with the primary and secondary amine groups of drugs to undergo degradation via the Maillard reaction. This topic will be covered in Chapter 5, Drug–Excipient Interaction and Adduct Formation.
As indicated above, this book focuses on the organic chemistry of drug degradation, in particular, the mechanisms and pathways of the chemical degradation of both small and large molecule drugs under real life degradation scenarios. Owing to the variety of dosage forms of formulated drugs, degradation of drugs can occur in various states including solid (tablets, capsules, and powders), semi-solid (creams, o...
Table of contents
- Cover image
- Title Page
- Copyright
- Preface
- Contents
- Chapter 1 Introduction
- Chapter 2 Hydrolytic Degradation
- Chapter 3 Oxidative Degradation
- Chapter 4 Various Types and Mechanisms of Degradation Reactions
- Chapter 5 Drug–Excipient Interactions and Adduct Formation
- Chapter 6 Photochemical Degradation
- Chapter 7 Chemical Degradation of Biological Drugs
- Chapter 8 Strategies for Elucidation of Degradant Structures and Degradation Pathways
- Chapter 9 Control of Drug Degradation
- Subject Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Organic Chemistry of Drug Degradation by Min Li in PDF and/or ePUB format, as well as other popular books in Medicina & Farmacologia. We have over 1.5 million books available in our catalogue for you to explore.