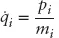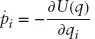INTRODUCTION
Molecular dynamics (MD) is a powerful tool in modern chemistry that allows one to describe the time evolution of a computational model for a complex molecular system.1–3 Typical models range from being highly accurate where energy and forces are computed with advanced and expensive quantum chemistry methods to faster but less accurate empirically parameterized force fields at atomistic or coarser resolution. The power of these techniques lies in their ability to reproduce experimental observable quantities accurately while, at the same time, giving access to the mechanistic details of chemical reactions or conformational changes at very high spatial resolution - typically at atomistic scale. For this reason, MD is often used to complement experimental investigations and to help in interpreting experiments and in designing new ones. Moreover, thanks to new parallelization algorithms and to the continuous improvements in computer hardware driven by Moore’s law, the range of application of these techniques has grown exponentially in the past decades and can be expected to continue growing.
In spite of its success, however, MD is still limited to the study of events on a very short timescale. Indeed, depending on the required accuracy and on the available computational resources, MD can provide trajectories for events happening on the timescale of picoseconds (quantum chemistry) to microseconds (empirical force fields). Thus, many interesting phenomena, namely, chemical reactions, protein folding and aggregation, and macromolecular rearrangement are still out of reach of direct investigation using straightforward MD trajectories. Besides the optimization of computer software (e.g., Ref. 4) and/or hardware (e.g. Refs. 5, 6), it is a possible complementary strategy to alleviate this issue by using algorithms where the time evolution is modified to sample more frequently the event under investigation. Then, appropriate postprocessing techniques are necessary to recover unbiased properties from the accelerated trajectories.
Many algorithms to accelerate MD simulations have been designed in the past decades, and a discussion of all of them is out of the scope of this chapter. Some of these algorithms are based on increasing the temperature of the simulated system (e.g., parallel tempering7 and solute tempering8), while others are based on exploiting an a priori knowledge of the investigated transition to design a proper order parameter to both describe and accelerate it. This last class includes umbrella sampling,9 adaptive biasing force,10 metadynamics,11 self-healing umbrella sampling,12 and other methods that keep the selected order parameters at an artificially high temperature.13–15 This chapter focuses on metadynamics, which was first introduced in 200211 and then improved with several variants in the past decade. Metadynamics has been employed successfully in several fields, ranging from chemical reactions16 to protein folding17 and aggregation,18 molecular docking,19 crystal structure prediction,20 and nucleation.21 A further push in the diffusion of metadynamics application has been its availability in a few widespread molecular dynamics codes22–24 and in open-source plugins.25–27
The main goal of this chapter is to provide an entry-level tutorial for metadynamics. In Section “Molecular Dynamics and Free-Energy Estimation” we provide an introduction to the basic concepts of molecular dynamics and of free-energy calculations. In Section “A Toy Model: Alanine Dipeptide” we introduce a toy model that will then be used for subsequent examples. Section “Biased Sampling” is devoted to the introduction of biased sampling. In Sections “Adaptive Biasing with Metadynamics” and “Well-Tempered Metadynamics” metadynamics is introduced, and Section “Metadynamics How-To” provides a practical how-to for performing a free-energy calculation with metadynamics. For all the simulations described in that section a sample input file for the open-source package PLUMED 226 is given in the Appendix. In the remaining sections, a quick overview of some of the latest improvements in the field is given, followed by a concluding section.