Neurodevelopmental disorders arise from disturbances to various processes of brain development, which can manifest in diverse ways. They encompass many rare genetic syndromes as well as common, heritable conditions such as intellectual disability, autism, ADHD, schizophrenia and many types of epilepsy. The Genetics of Neurodevelopmental Disorders examines recent revolutionary advances in our understanding of the genetics of these disorders, exploring both basic discoveries and the translation of new findings into the clinical setting.
The book begins by examining the genetic architecture and etiology of neurodevelopmental disorders. It describes the striking recent progress in identifying pathogenic mutations, which are grouped here based on the neurodevelopmental processes impacted. Subsequent chapters consider the use of cellular and animal models to elucidate the cascading consequences of such mutations, from molecular and cellular levels to emergent effects on neural circuits, brain systems and subsequent psychological development. The text concludes by examining the important clinical implications of the recent advances in the field, from recognition of the genetic causes in individual patients to development of new treatments and interventions.
A timely synthesis, The Genetics of Neurodevelopmental Disorders is a unique and essential resource for neuroscientists, geneticists, neurologists and psychiatrists and an accessible and up-to-date overview for medical and science students.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Chapter 1 The Genetic Architecture of Neurodevelopmental Disorders
Kevin J. Mitchell
Smurfit Institute of Genetics and Institute of Neuroscience, Trinity College Dublin, Dublin, Ireland
1.1 Introduction
There are several hundred known genetic syndromes that affect neural development and result in intellectual disability (ID), epilepsy, or other neurological or psychiatric symptoms. These include recognized syndromes that often manifest with symptoms of autism spectrum disorders (ASD) or schizophrenia (SZ), such as Fragile X syndrome, Rett syndrome, tuberous sclerosis, velocardio-facial syndrome, and many others. For ASD, it has been known for many years that these syndromes account for a significant but still small fraction (5–10%) of all cases (Miles, 2011). What has not been clear is whether such cases, associated with single mutations, represent a typical mode by which such conditions arise or are, alternatively, exceptional and quite distinct from the general etiology of idiopathic ASD, epilepsy, SZ, or ID (Wray and Visscher, 2010). Other common disorders including dyslexia, specific language impairment, obessive-compulsive disorder, and so on, will not be considered here in detail, though the general principles probably apply.
In general, the genetic architecture of common NDDs has been considered to be “complex” or multifactorial (Plomin et al., 2009; Sullivan et al., 2003). This is usually taken to mean that many causal factors, both genetic and nongenetic, are involved in each affected individual. Under this view, the large group of currently idiopathic cases have a very different genetic architecture from the small number of known monogenic cases. An alternative view is that the vast majority of cases of these conditions are caused by independent mutations in any one of a very large number of genes. According to this model, these diagnostic categories of idiopathic cases represent artificial groupings reflecting our current ignorance, rather than natural kinds.
Here, I consider the theoretical underpinnings and empirical evidence relating to the genetic architecture of NDDs. These have been greatly influenced by technological advancements which have allowed various types of genetic variation to be assayed. Studies over the past several years have revealed an extreme level of genetic heterogeneity and complexity for common NDDs, with the discovery of high-risk mutations in a large number of single loci and additional complexities in the causal architecture in individuals.
1.2 Theoretical Considerations
Linkage studies have clearly shown that common NDDs are not caused by mutations in one particular gene, leading to the unchallenged conclusion that variants at many loci must be involved across the population (e.g., (O'Rourke et al., 1982; Szatmari, 1999)). However, models of the genetic architecture of these conditions differ in two additional, independent parameters: (i) the number of variants thought to contribute to disease in any individual (from one or a few to many, possibly thousands), and (ii) the presumed frequency of risk alleles (from very rare to very common). The differences between models have profound implications for finding causal variants, predicting disease risk, discovering underlying biology, and developing treatments for particular patients. More fundamentally, they represent very different ways of conceptualizing these conditions.
1.2.1 Number of Causal Alleles Per Individual
At one extreme, a model of Mendelian inheritance with genetic heterogeneity proposes that each case is caused by a single mutation, but that these can occur in any one of a large number of different loci (McClellan and King, 2010; Mitchell, 2011; Wright and Hastie, 2001). The types of mutations could include chromosomal aberrations that change the copy number of multiple genes, or mutations affecting a single gene. This model also encompasses diverse modes of inheritance, from de novo mutations to dominant or recessive inheritance. Fundamentally, this model conceives of common clinical categories such as SZ, ASD, epilepsy, ID, and so on, as umbrella terms for large numbers of distinct conditions that happen to manifest with similar symptoms (Betancur, 2011; McClellan et al., 2007; Mitchell, 2012; Mitchell and Porteous, 2011; Ropers, 2008).
There are many precedents for this kind of genetic heterogeneity, including the genetics of congenital deafness (Lenz and Avraham, 2011), various forms of blindness, such as retinitis pigmentosa (Wright et al., 2010) and the many known Mendelian forms of intellectual disability (Ellison et al., 2013) and epilepsy (Poduri and Lowenstein, 2011). What differs with these conditions is that they typically display clear-cut Mendelian modes of inheritance, which is rarely the case for NDDs.
Moreover, linkage studies have been highly successful in identifying causal loci involved in specific Mendelian sub-types of these disorders, whereas they have produced highly inconsistent findings for common diagnostic categories, such as ASD and SZ (see below). Partly due to the failure of linkage studies to zero in on specific causal loci, an alternative model of polygenic inheritance became the dominant paradigm in the field (Risch, 1990).
The polygenic model proposes that common disorders arise from the combined action of a large number of risk alleles in each affected individual (Falconer, 1965; Plomin et al., 2009). Regrettably, the term polygenic has been used more loosely in recent literature to refer simply to the involvement of many loci across the population, where the number of contributing loci per individual remains unknown and could be as low as one (Purcell et al., 2014; Sullivan et al., 2012). I use polygenic here in the original sense to refer to conditions caused by the combined effects of multiple variants per individual.
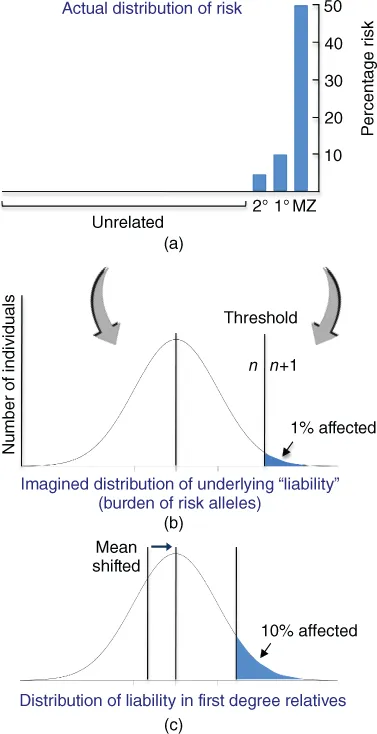
Under the polygenic model, many risk variants are floating through the population and their independent segregation generates a continuous distribution of risk variant burden. Individuals at the extreme end of this distribution are thought of as passing a threshold and consequently developing disease (Falconer, 1965) (Fig. 1.1). This model views common disorders effectively as unitary conditions, reflecting a common etiology – people with disease are simply at the tail end of a single distribution that extends continuously across the whole population. The distribution in this case is of the imagined latent variable, “liability,” which is presumed to exist and to be normally distributed, but which cannot be measured directly. It can be translated, statistically, into the highly discontinuous distribution of observed risk in relatives of affected individuals, for example, by invoking an essentially arbitrary threshold, above which disease results. This liability-threshold model is statistically convenient but highly abstract (Mitchell, 2012).
Figure 1.1The liability-threshold model. A discontinuous distribution of observed risk across the population (a) is represented as reflecting an underlying latent variable, the “liability”, which is assumed to be normally distributed (b). A threshold of burden is invoked to regenerate the observed discontinuity. The mean liability of siblings of affected individuals is presumed to be shifted toward the threshold (c), explaining the greater disease incidence in this group compared to the population average. This yields a scenario analogous to response to selection for a quantitative trait, enabling heritability to be estimated (Falconer, 1965).
(Reproduced, with permission, from (Mitchell, 2012).)
An extension of this model considers the disorder as arising from the extremes of a number of actual quantitative traits, or endophenotypes (Gottesman and Gould, 2003; Meyer-Lindenberg and Weinberger, 2006). Common neuropsychiatric conditions affect multiple cognitive or social functions or faculties, such as working memory, executive function, sociability, and so on. All of these traits also show a distribution across the unaffected population and all show moderate heritability. This led to the suggestion that individuals diagnosed with conditions such as ASD or SZ may simply be at the extreme end of the normal distributions for several of these traits at the same time (Gottesman and Gould, 2003; Meyer-Lindenberg and Weinberger, 2006).
The corollary of that idea is that the genetic variants contributing to variation in such traits across the normal population will be the risk variants for such disorders. The hope was that such traits might have simpler genetic architectures than clinical diagnoses or at least that any genetic associations would be more obvious, as these traits reflect functions supposedly closer to the action of the genes.
1.2.2 Frequency of Risk Alleles – Evolutionary Considerations
In addition to the number of loci involved, the ...
Table of contents
Cover
Table of Contents
Foreword
Chapter 1: The Genetic Architecture of Neurodevelopmental Disorders
Chapter 2: Overlapping Etiology of Neurodevelopmental Disorders
Chapter 3: The Mutational Spectrum of Neurodevelopmental Disorders
Chapter 4: The Role of Genetic Interactions in Neurodevelopmental Disorders
Chapter 5: Developmental Instability, Mutation Load, and Neurodevelopmental Disorders
Chapter 6: Environmental Factors and Gene–Environment Interactions
Chapter 7: The Genetics of Brain Malformations
Chapter 8: Disorders of Axon Guidance
Chapter 9: Synaptic Disorders
Chapter 10: Human Stem Cell Models of Neurodevelopmental Disorders
Chapter 11: Animal Models for Neurodevelopmental Disorders
Chapter 12: Cascading Genetic and Environmental Effects on Development: Implications for Intervention
Chapter 13: Human Genetics and Clinical Aspects of Neurodevelopmental Disorders
Chapter 14: Progress Toward Therapies and Interventions for Neurodevelopmental Disorders
Subject Index
Gene Index
End User License Agreement
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access The Genetics of Neurodevelopmental Disorders by Kevin J. Mitchell in PDF and/or ePUB format, as well as other popular books in Biological Sciences & Neuroscience. We have over 1.5 million books available in our catalogue for you to explore.