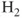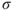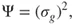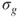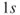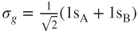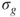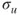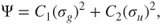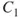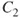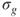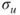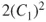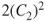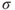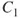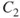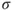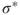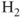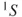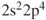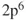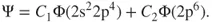The first book to aid in the understanding of multiconfigurational quantum chemistry, Multiconfigurational Quantum Chemistry demystifies a subject that has historically been considered difficult to learn. Accessible to any reader with a background in quantum mechanics and quantum chemistry, the book contains illustrative examples showing how these methods can be used in various areas of chemistry, such as chemical reactions in ground and excited states, transition metal and other heavy element systems. The authors detail the drawbacks and limitations of DFT and coupled-cluster based methods and offer alternative, wavefunction-based methods more suitable for smaller molecules.
How do we define multiconfigurational (MC) methods? It is simple. In Hartree–Fock (HF) theory and density functional theory (DFT), we describe the wave function with a single Slater determinant. Multiconfigurational wave functions, on the other hand, are constructed as a linear combination of several determinants, or configuration state functions (CSFs)—each CSF is a spin-adapted linear combination of determinants. The MC wave functions also go by the name Configuration Interaction (CI) wave function. A simple example illustrates the situation. The
molecule (centers denoted A and B) equilibrium is well described by a single determinant with a doubly occupied
orbital:
1.1
where
is the symmetric combination of the
atomic hydrogen orbitals (
; the antisymmetric combination is denoted as
). However, if we let the distance between the two atoms increase, the situation becomes more complex. The true wave function for two separated atoms is
1.2
which translates to the electronic structure of the homolytic dissociation products of two radical hydrogens. Two configurations,
and
, are now needed to describe the electronic structure. It is not difficult to understand that at intermediate distances the wave function will vary from Eq. 1.1 to Eq. 1.2, a situation that we can describe with the following wave function:
1.3
where
and
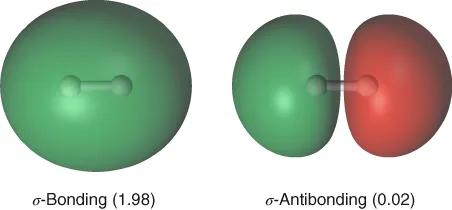
, the so-called CI-coefficients or expansion coefficients, are determined variationally. The two orbitals,
and
, are shown in Figure 1.1, which also gives the occupation numbers (computed as
and
) at a geometry close to equilibrium. In general, Eq. 1.3 facilitates the description of the electronic structure during any
bond dissociation, be it homolytic, ionic, or a combination of the two, by adjusting the variational parameters
and
accordingly.
Figure 1.1 The
and
orbitals and associated occupation numbers in the
molecule at the equilibrium geometry.
This little example describes the essence of multiconfigurational quantum chemistry. By introducing several CSFs in the expansion of the wave function, we can describe the electronic structure for a more general situation than those where the wave function is dominated by a single determinant. Optimizing the orbitals and the expansion coefficients, simultaneously, defines the approach and results in a wave function that is qualitatively correct for the problem we are studying (e.g., the dissociation of a chemical bond as the example above illustrates). It remains to describe the effect of dynamic electron correlation, which is not more included in this approach than it is in the HF method.
The MC approach is almost as old as quantum chemistry itself. Maybe one could consider the Heitler–London wave function [1] as the first multiconfigurational wave function because it can be written in the form given by Eq. 1.2. However, the first multiconfigurational (MC) SCF calculation was probably performed by Hartree and coworkers [2]. They realized that for the
state of the oxygen atom, there where two possible configurations,
and
, and constructed the two configurational wave function:
1.4
The atomic orbitals were determined (numerically) together with the two expansion coefficients. Similar MCSCF calculations on atoms and negative ions were simultaneously performed in Kaunas, Lithuania, by Jucys [3]. The possibility was actually suggested already in 1934 in the book by Frenkel [4]. Further progress was only possible with the advent of the computer. Wahl and Das developed the Optimized Valence Configuration (OVC) Approach, which was applied to diatomic and some triatomic molecules [5, 6].
An important methodological step forward was the formulation of the Extended Brillouin's (Brillouin, Levy, Berthier) theorem by Levy and Berthier [7]. This theor...
Table of contents
Cover
Title Page
Copyright
Dedication
Table of Contents
Preface
Conventions and Units
Chapter 1: Introduction
Chapter 2: Mathematical Background
Chapter 3: Molecular Orbital Theory
Chapter 4: Hartree–Fock Theory
Chapter 5: Relativistic Effects
Chapter 6: Basis Sets
Chapter 7: Second quantization and multiconfigurational wave functions
Chapter 8: Electron correlation
Chapter 9: Multiconfigurational SCF Theory
Chapter 10: The RAS State-Interaction method
Chapter 11: The Multireference CI Method
Chapter 12: Multiconfigurational Reference Perturbation Theory
Chapter 13: CASPT2/CASSCF Applications
Summary and Conclusion
Index
End User License Agreement
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Multiconfigurational Quantum Chemistry by Björn O. Roos,Roland Lindh,Per Åke Malmqvist,Valera Veryazov,Per-Olof Widmark,Per �ke Malmqvist in PDF and/or ePUB format, as well as other popular books in Physical Sciences & Physical & Theoretical Chemistry. We have over one million books available in our catalogue for you to explore.