Chemistry
Nucleophilic Substitution Reactions
Nucleophilic substitution reactions involve the replacement of a leaving group in a molecule with a nucleophile. This process occurs when a nucleophile attacks an electrophilic center, leading to the formation of a new bond and the displacement of the leaving group. These reactions are important in organic chemistry and are commonly used in the synthesis of various organic compounds.
Written by Perlego with AI-assistance
Related key terms
1 of 5
12 Key excerpts on "Nucleophilic Substitution Reactions"
 eBook - ePub
eBook - ePubEssentials of Organic Chemistry
For Students of Pharmacy, Medicinal Chemistry and Biological Chemistry
- Paul M. Dewick(Author)
- 2013(Publication Date)
- Wiley(Publisher)
6
Nucleophilic reactions: nucleophilic substitution
As the term suggests, a substitution reaction is one in which one group is substituted for another. For nucleophilic substitution, the reagent is a suitable nucleophile and it displaces a leaving group. As we study the reactions further, we shall see that mechanistically related competing reactions, eliminations and rearrangements, also need to be considered.6.1 The SN 2 reaction: bimolecular nucleophilic substitution
The abbreviation SN 2 conveys the information ‘substitution–nucleophilic–bimolecular’. The reaction is essentially the displacement of one group, a leaving group, by another group, a nucleophile. It is a bimolecular reaction, since kinetic data indicate that two species are involved in the rate-determining step:where Nu is the nucleophile, RL the substrate containing the leaving group L, and k is the rate constant.In general terms, the reaction can be represented as belowDifferences in electronegativities (see Section 2.7) between carbon and the leaving group atom lead to bond polarity. This confers a partial positive charge on the carbon and facilitates attack of the nucleophile. As the nucleophile electrons are used to make a new bond to the carbon, electrons must be transferred away to a suitable acceptor in order to maintain carbon’s octet. The suitable acceptor is the electronegative leaving group.The nucleophile attacks from the side opposite the leaving group – electrostatic repulsion prevents attack in the region of the leaving group. This results in an inversion process for the other groups on the carbon centre under attack, rather like an umbrella turning inside out in a violent gust of wind. The process is concerted, i.e. the bond to the incoming nucleophile is made at the same time as the bond to the leaving group is being broken. As a consequence, the mechanism involves a high-energy transition state in which both nucleophile and leaving group are partially bonded, the Nu–C–L bonding is linear, and the three groups X, Y, and Z around carbon are in a planar array. This is the natural arrangement to minimize steric interactions if we wish to position five groups around an atom, and will involve three sp 2 orbitals and a p orbital as shown. The p orbital is used for the partial bonding; note that we cannot have five full bonds to a carbon atom. The energy profile for the reaction (Figure 6.1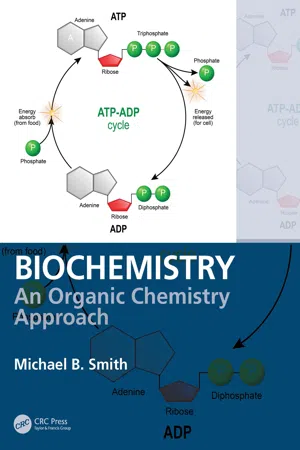 eBook - ePub
eBook - ePubBiochemistry
An Organic Chemistry Approach
- Michael B. Smith(Author)
- 2020(Publication Date)
- CRC Press(Publisher)
3 Nucleophiles and ElectrophilesAliphatic substitution reactions are early examples of organic chemical reactions in a typical undergraduate organic chemistry course. Such reactions involve the reaction of nucleophilic species with an electrophilic species, and for the most part they follow first-order or second-order kinetics. There are nucleophiles that are prevalent in biochemical reactions, including alcohols, amines, and thiols. Substitution reactions in a typical organic chemistry course involve reactions at carbon that is connected to a heteroatom moiety such as a halogen leaving group. In biochemistry the leaving group is often a phosphonate ester or another biocompatible group. Another type of nucleophilic reaction involves carbonyl compounds, including acyl addition of ketone and aldehyde moieties and acyl substitution reactions of carboxylic acid derivatives.This chapter will briefly review the SN 2 and SN 1 reactions and then describe nucleophiles that are common in biochemical applications and the substitution reactions that are common for these nucleophiles. Nucleophilic reactions require electrophilic species. Electrophiles or electrophilic substrates are common in biochemistry, including phosphonate derivatives, carbonyl compounds and imine compounds. Any discussion of typical nucleophilic reactions also requires an understanding of such electrophilic substrates. The fundamentals of both acyl addition and of acyl substitution reactions will be presented for carbonyl electrophilic centers and the reactions of these electrophilic centers with nucleophiles.3.1 Nucleophiles and Bimolecular Substitution (the SN 2 Reaction)The SN 2 reaction is one of the seminal reactions in a typical undergraduate organic chemistry course. The reaction of 1-bromo-3-methylbutane with sodium iodide (NaI) using acetone as a solvent gave 1-iodo-3-methylbutane, in 66% yield.1 In terms of the structural changes, the iodide ion substitutes for the bromine, producing bromide ion (Br– ). Iodide reacted as a nucleophile in the reaction at Cδ+ of the alkyl bromide, breaking the C—Br bond and transferring the electrons in that bond to bromine. In molecules that contain the C—Br bond, or indeed a C—C bond, where X is a heteroatom-containing group, the carbon will have a δ+ dipole. In other words, the carbon atom is electrophilic, and the substrate that reacts with the nucleophile is called an electrophile. The reaction of a nucleophile with an aliphatic electrophile is formally called nucleophilic aliphatic substitution , illustrated in Figure 3.1 . The displaced atom or group (e.g., chloride), departs (leaves) to become an independent ion. Displacement of chlorine leads to the chloride ion (Cl– ), but the bromide ion, iodide ion, or a sulfonate anion also correlates to X, which is referred to as a leaving group . In many biochemical reactions, the leaving group is a phosphate, —O–PO2 eBook - PDF
eBook - PDFOrganic Reaction Mechanisms 2018
An Annual Survey Covering the Literature Dated January to December 2018
- Mark G. Moloney(Author)
- 2021(Publication Date)
- Wiley(Publisher)
247 7 Nucleophilic Aliphatic Substitution 2018 J. G. Moloney and M. G. Moloney Department of Chemistry, University of Oxford, Oxford, UK CHAPTER MENU General, 247 S N 1 Mechanistic Studies, 248 S N 2 Mechanistic Studies, 249 S N – General Mechanistic Studies, 251 Nucleophiles, 254 Fluorination, 254 Propargyl Substitutions, 255 Glycosylations, 255 Reactions, 256 Boron-mediated Processes, 264 Iodine-mediated Processes, 264 Metal-catalysed Processes, 264 Photomediated Processes, 267 Nucleophilic Substitutions at Heteroatoms, 268 Other Reactions, 270 References, 272 General A study of organic chemistry students’ ability to match one of three possible reaction coordinate diagrams with a nucleophilic substitution reaction mechanism has shown that they do not read-ily see the connection of a representation of a reaction with the physical reality of that reaction, that is, that the process proceeds by the collisions of atoms and molecules of appropriate energy. Moreover, the tendency of expert organic chemistry tutors to write reaction equations which are balanced neither in charge nor mass, concentrating instead on important species (reactant, intermediate, and product), and ignoring other participating reaction species (leaving groups, nucleophiles/bases and solvent molecules) exacerbates this misunderstanding. It is suggested that symbolism needs careful explanation and development, and its link to submicroscopic and macroscopic domains constantly emphasized. 1 Furthermore, understanding of leaving-group ability seems to be superficial, notwithstanding the well-known relationship that good leav-ing groups are the weak conjugate bases of strong acids. This seems to arise because students find the prediction and rationalization of relative acid strength to be difficult because of the need to invoke multiple concepts simultaneously (e.g. atomic size, electronegativity, electronic effects (resonance and induction), orbital hybridization). eBook - PDF
eBook - PDF- T. W. Graham Solomons, Craig B. Fryhle, Scott A. Snyder(Authors)
- 2017(Publication Date)
- Wiley(Publisher)
(c) Br (b) CH 3 Br (a) 6.2 Nucleophilic Substitution Reactions Nucleophilic Substitution Reactions are among the most fundamental types of organic reactions. In a nucleophilic substitution reaction a nucleophile (Nu ⋅ ⋅ ) displaces a leaving group (LG) in the molecule that undergoes the substitution (the substrate). • The nucleophile is always a Lewis base (electron pair donor), and it may be negatively charged or neutral. • The leaving group is always a species that takes a pair of electrons with it when it departs. Often the substrate is an alkyl halide ( R − X ⋅⋅ ⋅⋅ ⋅ ⋅ ) and the leaving group is a halide anion ( ⋅ ⋅ X ⋅⋅ ⋅⋅ ⋅ ⋅ − ). The following equations include a generic nucleophilic substitution reaction and some specific examples. PRACTICE PROBLEM 6.2 Classify each of the following organic halides as primary, secondary, tertiary, alkenyl, or aryl. (e) I (d) F (c) Br (b) Cl (a) Br 6.2 Nucleophilic Substitution Reactions 243 + + + OCH 3 CH 3 CH 2 CH 3 OH Nu R The nucleophile uses its electron pair to form a new covalent bond with the substrate carbon. + + + − CH 3 O − HO − Nu The nucleophile is a Lewis base that donates an electron pair to the substrate. Br − I − LG − The leaving group gains the pair of electrons that originally bonded it in the substrate. CH 3 CH 2 Br CH 3 I LG R The bond between the carbon and the leaving group breaks, giving both electrons from the bond to the leaving group. + I + − Cl Cl I − + + R— N—CH 3 R R + R—N R R I − H 3 C—I In Nucleophilic Substitution Reactions the bond between the substrate carbon and the leaving group undergoes heterolytic bond cleavage. The unshared electron pair of the nucleophile forms the new bond to the carbon atom. eBook - ePub
eBook - ePub- Robert J. Ouellette, J. David Rawn(Authors)
- 2015(Publication Date)
- Elsevier(Publisher)
Most mollusks are protected from predators by a hard shell, so the shell-less sea hare might seem to have little prospect of survival in the face of large carnivores. However, the sea hare converts the halogen-containing compounds in red algae into closely related substances and secretes them in a mucous coating. This coating protects its soft body against carnivorous fish, which are repelled by the compounds.7.2 Nucleophilic Substitution Reactions
In a nucleophilic substitution reaction, the nucleophile donates an electron pair to the electrophilic carbon atom to form a carbon-nucleophile bond. The nucleophile reacts with a haloalkane, which is called the substrate; that is, the compound upon which the reaction occurs. The nucleophile may be either negatively charged, as in the case of OH− , or neutral, as in the case of NH3. These two types of nucleophiles are commonly represented as Nu:− and Nu:, respectively. If the nucleophile is negatively charged, the product has no net charge. If the nucleophile is neutral, the product is positively charged.1.2.The group displaced by the nucleophile is called the leaving group . It has an electron pair that was originally in the C—X bond. Haloalkanes can react with a halide anion, as in the case of the nucleophilic substitution of bromide for iodide in the substrate iodomethane.A similar reaction occurs when the hydroxide ion replaces a halide ion to produce an alcohol. When the oxygen-containing nucleophile is an alkoxide ion (RO− ), the product is an ether.Nucleophilic Substitution Reactions by sulfur-containing nucleophiles, such as hydrogen sulfide ion, HS− , and thiolate ions, RS− , also occur. These reactions yield sulfur analogs of alcohols and ethers— namely, thiols and thioethers (Chapter 8 ).Haloalkanes also react with carbon-containing nucleophiles to form carbon-carbon bonds, which increase the length of the carbon chain. The cyanide ion, C ≡ N− , is a carbon-containing nucleophile. In the reaction shown below it produces a nitrile with the formula RCN, which extends the carbon chain by one carbon atom. Nitriles can be transformed into carboxylic acids (Chapter 12 ) and amines (Chapter 14 ). Carbon-containing nucleophiles derived from alkynes are called alkynide ions. These nucleophiles, the conjugate bases of alkynes (Chapter 4 eBook - PDF
eBook - PDF- David R. Klein(Author)
- 2020(Publication Date)
- Wiley(Publisher)
These physiological responses prepare the body for “fight or flight.” 288 CHAPTER 7 Substitution Reactions Many substitution reactions appear to follow this stepwise mechanism. There are several pieces of evidence that support this stepwise mechanism in those cases. This evidence will now be explored. Kinetics Many substitution reactions do not exhibit second-order kinetics. Consider the following example: Br NaBr Na + + In the reaction above, the rate is dependent only on the concentration of the substrate. The rate equa- tion has the following form: Rate = k [substrate] Increasing or decreasing the concentration of the nucleophile has no measurable effect on the rate. The rate equation is said to be first order, because the rate is linearly dependent on the concentration of only one compound. In such cases, the mechanism must exhibit a rate-determining step in which the nucleophile does not participate. Because that step involves only one chemical entity, it is said to be unimolecular. Ingold and Hughes coined the term S N 1 to refer to unimolecular substitution reactions: S N Substitution Unimolecular Nucleophilic 1 When we use the term unimolecular, we don’t mean that the nucleophile is completely irrelevant. Clearly, the nucleophile is necessary, or there won’t be a reaction. The term unimolecular simply describes the fact that only one chemical entity participates in the rate-determining step of the reac- tion, and as a result, the rate of the reaction is not affected by how much nucleophile is present. That is, the rate of an S N 1 process is dependent only on the rate at which the leaving group leaves. As a result, the rate of an S N 1 process will only be affected by factors that affect the rate of that step. Increasing the concentration of the nucleophile has no impact on the rate at which the leaving group leaves. It is true that the nucleophile must be present in order to obtain the product, but an excess of nucleophile will not speed up the reaction. eBook - PDF
eBook - PDFPractical Synthetic Organic Chemistry
Reactions, Principles, and Techniques
- Stéphane Caron(Author)
- 2020(Publication Date)
- Wiley(Publisher)
231 4 Nucleophilic Aromatic Substitution Stéphane Caron and Emma McInturff Pfizer Worldwide R&D, Groton, CT, USA CHAPTER MENU Introduction, 231 Oxygen Nucleophiles, 232 Sulfur Nucleophiles, 234 Nitrogen Nucleophiles, 236 Halogen Nucleophiles, 241 Carbon Nucleophiles, 243 ortho-Arynes, 245 4.1 Introduction Nucleophilic aromatic substitution (S N Ar), which can operate through several different reaction mechanisms, is considered one of the preferred methods to derivatize arenes. As such, there are numerous examples of simple functionalization and complex fragment union. Despite great advances in transition metal-catalyzed arene function- alization, S N Ar remains an attractive option due to simplicity, low cost, and avoidance of metal contamination of the product. The scope of this reaction is guided by three basic principles: electron deficiency at the reactive carbon on the aromatic system, nature of the leaving group to be displaced, and reactivity of the nucleophile. 1 In general, more electron-deficient arenes will undergo more facile aromatic nucleophilic substitution in an addition/elimination sequence. Aryl halides, specifically fluorides, and diazonium compounds have proven to be the most successful substrates for this reaction. While the typical order of reactivity for an aliphatic nucleophilic substitution follows I − > Br − > Cl − ≫ F − , this trend is generally reversed for the nucleophilic aromatic substitution. The electron with- drawing nature of an aryl fluoride enhances the propensity for nucleophilic attack at the fluorine-bearing carbon. Primary and secondary amines, as well as alkoxides, are usually excellent nucleophiles for the reaction. A few types of carbon nucleophiles, including cyanide and malonate derivatives, are also commonly used. The preparation of ortho-arynes will also be briefly discussed in this chapter. eBook - ePub
eBook - ePub- V. Ramamurthy, Kirk S. Schanze(Authors)
- 2005(Publication Date)
- CRC Press(Publisher)
4 PhotoNucleophilic Substitution ReactionsMaurizio Fagnoni and Angelo Albini
CONTENTS 4.1 Introduction 4.2 Mechanism 4.3 Formation of a C-C or Other C-IV Group Elements Bond4.3.1 Alkylation of Aryl Halides via the SN 1 Path4.3.2 Alkylation of Aryl Halides via the SRN 1 Path4.3.3 Alkylation of Aryl Nitriles 4.3.4 Cyanation and Carboxylation 4.3.5 Formation of a C-Si Bond 4.3.6 Formation of a C-Sn Bond 4.4 Formation of a C-V Group Elements Bond 4.4.1 Formation of Amines and Imines 4.4.2 Formation of Nitro Compounds 4.4.3 Formation of a C-P Bond 4.4.4 Formation of a C-As and C-Sb Bond 4.5 Formation of a C-VI Group Elements Bond, Except Oxygen 4.5.1 Formation of a C-S Bond 4.5.2 Formation of a C-Se or C-Te Bond 4.6 Formation of a C-Halogen Bond 4.7 Formation of a C-O Bond 4.8 Synthetic Significance of Photonucleophilic Substitutions4.1 INTRODUCTION
Nucleophilic Substitution Reactions form a significant chapter in the photochemistry of aromatics. Interest in the topic started with the observation in Havinga laboratory in 1956 that the hydrolysis of nitrophenyl phosphates and sulfates, not occurring to a significant extent at room temperature, was much accelerated by (room) light and actually occurred smoothly under irradiation, in particular in the case of the meta isomers [1 ]. A striking point in this reaction is the “meta activation” by the nitro group, in contrast with the ortho-para activation characteristic of thermal nucleophilic substitutions. Along with the opposite stereochemical course of thermal and photochemical cyclization reactions in Vitamin D derivatives, reported shortly afterwards by the same group [2 ], this was one of the key observations evidencing the different behavior of excited states vs. ground states and gave impetus to the development of organic photochemistry and its rationalization in the following years (see, e.g., Refs. 3 ,4 ,5 eBook - PDF
eBook - PDF- Metin Balcı(Author)
- 2021(Publication Date)
- Wiley-VCH(Publisher)
However, the situation is somewhat dif- ferent in S N 2 ′ reactions. The nucleophile approaches the double bond on the side from which the leaving group departs, which is called a syn-attack. In such an attack, double-bond electrons open backward to remove the X-group. Thus, the electron density is increased at the back of the leaving group. Substitution occurs by the attack of electrons on the car- bon atom to which the leaving group is bonded. If the nucleophile attacks the double bond from the opposite direction (anti-attack), the electron density will increase at the side of the leaving group, which will not be suitable for a substitution reaction. 74 2 Nucleophilic Substitution Reaction C CH C X Nu syn-attack C HC C Nu The syn-attack can be nicely demonstrated in cyclic structures. In the example given below, the nucleophile attacks the molecule from the side of the leaving group and removes benzoate [19]. C(CH 3 ) 3 O O Ph N H C(CH 3 ) 3 N 2.3.4 Internal Nucleophilic Substitution Reaction, S N i In the previous sections, we have discussed S N 1 and S N 2 mechanisms. S N 2 reactions proceed through the formation of a transition state, resulting in configuration inversion of the product. There are still other reactions whose stereochemical outcome cannot be explained by S N 1 or S N 2 mechanisms. In some Nucleophilic Substitution Reactions, although the reaction molecularity is bimolecular, retention of the configuration is observed instead of inversion. These and similar reactions are often observed by the reaction of chiral alcohol with thionyl chloride to give the corresponding alkyl halide. In the first step, the oxygen atom of alcohol attacks the sulfur atom of thionyl chloride and removes one of the chlorine atoms attached to the sulfur atom to form alkyl chlorosulfite, which can be isolated. At this stage, there is no configurational change at the chiral carbon atom as the nucleophilic substitution reaction takes place on the sulfur atom. eBook - PDF
eBook - PDF- David R. Klein(Author)
- 2021(Publication Date)
- Wiley(Publisher)
332 CHAPTER 7 Alkyl Halides: Nucleophilic Substitution and Elimination Reactions In order to propose a synthesis, it is certainly important to know what reactions are available, but more importantly, you must know how and when to use those reactions. When proposing a syn- thesis (even a one-step synthesis), it is generally helpful to think backwards. To illustrate what this means, consider the following S N 2 reaction, in which an alkyl halide is converted into an ether: An ether An alkyl halide Synthesis: Br O NaOEt This reaction can also be viewed backwards, like this: An ether An alkyl halide Br NaOEt can be made from + An alkoxide ion O Retrosynthesis: This figure represents a retrosynthetic analysis, in which the product is shown first, followed by reagents that can be used to make that product. The wavy line, also called a disconnection, identifies the bond that can be made by the reaction. The special arrow indicates a retrosynthesis, which means that we are think- ing about this reaction backwards. Planning a retrosynthesis requires that we identify a suitable nucleo- phile and electrophile that will react with each other to give the target molecule (the desired product). Considering the ether target molecule above, are there any other reasonable disconnections that can be made? By focusing on the bond(s) adjacent to the functional group, we can identify suitable nuc- leophiles that could be used in a substitution reaction. The bond indicated below represents another logical disconnection, because we know that oxygen atoms can be good nucleophiles. O What starting materials are needed? + ? ? With this disconnection, what would the starting materials look like? In order to form the indicated bond, one of the two atoms must have started out as a nucleophile, and the other must have started out as an electrophile. eBook - PDF
eBook - PDF- T. W. Graham Solomons, Craig B. Fryhle, Scott A. Snyder(Authors)
- 2016(Publication Date)
- Wiley(Publisher)
6.14 ORGANIC SYNTHESIS 273 Alkyl chlorides and bromides are also easily converted to alkyl iodides by Nucleophilic Substitution Reactions. or R Cl R Br R I ( + Cl or Br ) I - - - One other aspect of the S N 2 reaction that is of great importance is stereochemistry (Section 6.8). S N 2 reactions always occur with inversion of configuration at the atom that bears the leaving group. This means that when we use S N 2 reactions in syntheses we can be sure of the configuration of our product if we know the configuration of our reactant. For example, suppose we need a sample of the following nitrile with the (S ) configuration: N C CH 3 H CH 2 CH 3 C (S)-2-Methylbutanenitrile If we have available (R )-2-bromobutane, we can carry out the following synthesis: - Br + H CH 3 CH 2 CH 3 C (S)-2-Methylbutanenitrile C N (R)-2-Bromobutane H 3 CH 2 C H H 3 C (inversion) C S N 2 Br + - C N PRACTICE PROBLEM 6.19 Starting with (S )-2-bromobutane, outline syntheses of each of the following compounds: (c) SH ( R )-CH 3 CHCH 2 CH 3 (a) OCH 2 CH 3 ( R )-CH 3 CHCH 2 CH 3 (d) SCH 3 ( R )-CH 3 CHCH 2 CH 3 (b) OCCH 3 ( R)-CH 3 CHCH 2 CH 3 O THE CHEMISTRY OF... Biological Methylation: A Biological Nucleophilic Substitution Reaction The cells of living organisms synthesize many of the compounds they need from smaller molecules. Often these biosynthe- ses resemble the syntheses organic chemists carry out in their laboratories. Let us examine one example now. Many reactions taking place in the cells of plants and animals involve the transfer of a methyl group from an amino acid called methionine to some other compound. That this transfer takes place can be demonstrated experimentally by feeding a plant or animal methionine containing an isotopically labeled carbon atom (e.g., 13 C or 14 C) in its methyl group. Later, other compounds containing the “labeled” methyl group can be isolated from the organism. Some of the compounds that get their methyl groups from methionine are the following. eBook - PDF
eBook - PDF- T. W. Graham Solomons, Craig B. Fryhle, Scott A. Snyder(Authors)
- 2022(Publication Date)
- Wiley(Publisher)
• The relative strength of a nucleophile (its nucleophilicity) is measured in terms of the relative rate of its S N 2 reaction with a given substrate. A good nucleophile is one that reacts rapidly in an S N 2 reaction with a given substrate. A poor nucleophile is one that reacts slowly in an S N 2 reaction with the same substrate under comparable reaction conditions. (As mentioned above, we cannot compare nucleo- philicities with regard to S N 1 reactions because the nucleophile does not participate in the rate-determining step of an S N 1 reaction.) Methoxide anion, for example, is a good nucleophile for a substitution reaction with iodomethane. It reacts rapidly by an S N 2 mechanism to form dimethyl ether: CH 3 O⁻ + CH 3 I ⟶ CH 3 OCH 3 + I⁻ rapid PRACTICE PROBLEM 6.11 The relative rates of ethanolysis (solvolysis in ethanol) of four primary alkyl halides are as follows: CH 3 CH 2 Br, 1.0; CH 3 CH 2 CH 2 Br, 0.28; (CH 3 ) 2 CHCH 2 Br, 0.030; (CH 3 ) 3 CCH 2 Br, 0.00000042. (a) Is each of these reactions likely to be S N 1 or S N 2? (b) Provide an explanation for the relative reactivities that are observed. structure resembles the products. Conversely, in a highly exergonic step (red curve) the transition state lies close to the reactants in free energy, and we assume its structure resem- bles the reactants. The great value of the Hammond–Leffler postulate is that it gives us an intuitive way of visualizing those important, but fleeting, species that we call transition states. 6.13 Factors Affecting the Rates of S N 1 and S N 2 Reactions 271 272 CHAPTER 6 Nucleophilic Reactions Methanol, on the other hand, is a poor nucleophile for reaction with iodomethane.
Index pages curate the most relevant extracts from our library of academic textbooks. They’ve been created using an in-house natural language model (NLM), each adding context and meaning to key research topics.